
木质素化学催化解聚研究新进展
2017-08-07 05:50张学铭吴苗许凤
林业工程学报 2017年4期
张学铭,吴苗,许凤
(北京林业大学材料科学与技术学院,北京100083)
木质素化学催化解聚研究新进展
张学铭,吴苗,许凤*
(北京林业大学材料科学与技术学院,北京100083)
木质素作为木质纤维素三大组分之一,是自然界中芳香环含量最多的天然高分子聚合物。基于化学催化手段将其定向转化为化学品及材料,实现木质素高值转化及利用,替代不可再生的化石资源,已经成为国内外重大热门研究领域之一。为此,笔者对木质素化学催化降解转化方面的研究成果进行了综述,并对未来的研究方向进行了展望。基于不同催化反应机理,总结了催化还原和氧化降解体系,阐述了不同体系中使用的还原剂、氧化剂,对不同体系的反应机理及生成产物进行了阐述;同时,介绍了木质素化学降解常用的溶剂(水、有机溶剂、离子液体)和催化剂(液体酸、碱、固体酸、复合型催化剂)。总结认为,目前开发催化效率高、选择性好及低成本催化体系仍然是木质素定向催化转化研究的重点发展方向。
木质素;降解;催化剂;还原;氧化
利用生物质转化为高附加值的材料及化学品并替代不可再生的化石资源,已经成为全球重大热门研究课题之一,受到广泛关注[1]。生物质的主要组分包括纤维素(40%~50%)、半纤维素(25%~35%)和木质素(15%~20%)[2],三大组分通过分子内和分子间复杂的键合结构成复杂的木质纤维复合体。木质素是植物细胞壁的重要组成成分之一,是地球上最丰富的芳香族天然高分子化合物。它是由3种甲氧基化程度不同的4-羟基-肉桂醇(对香豆醇、松柏醇和芥子醇)经氧化聚合产生的天然高聚物[3-4],基本单元间连接键的类型主要包括β-O-4′,β-β′、β-5′等,含量最高的为β-O-4′连接键(45%~60%)[5]。同时,木质素分子含有众多种类的活性官能基,如羟基、甲氧基、羰基、羧基等,易被化学修饰,具有可再生、可降解、无毒等优点。因此,利用木质素可以生产许多潜在的高值产品,比如低成本的碳纤维[6]、工程塑料、热塑弹性体、聚合膜和泡沫[7],以及近些年备受关注的超级电容器[8]和芳香类化学品等[9]。然而,木质素自身结构单元的复杂性和成键机制多样性仍旧是阻碍木质素的高值化利用的关键障碍[10-11]。因此,针对木质素高值转化过程中存在的瓶颈问题,本文主要综述了木质素化学降解转化为化学品的研究进展。
利用化学法降解木质素,可以高得率、高选择性地得到目标芳香族产物[12]。从反应体系组成来分析,化学降解重点考虑溶剂和催化剂的选择,溶剂主要包括水、有机溶剂、离子液体三大溶剂体系;而催化剂从最初的液体酸、碱、固体酸,发展至目前常用的自行合成的复合型催化剂(如:有机/无机的金属复合催化剂等)。根据催化反应机理来划分,木质素化学催化降解的反应主要可以分为裂化、水解、催化还原和催化氧化反应[12]。在这四大反应中,由于还原和氧化反应的目标产物较易控制,因此受到更广泛关注。相比较而言,还原反应主要生成一些简单的芳香族化合物,而氧化反应趋向生成含有一些功能基的复杂化合物。因此,本文主要对溶剂与催化剂选择、还原和氧化过程、生成产物反应机理等方面进行分析。
1 木质素化学降解常用溶剂
在3类溶剂体系中,由于水作为最环保、经济的绿色溶剂深受研究者关注,因此,有关水热降解方面的研究最为广泛。根据处理的温度及压力差别,水热降解反应主要包括三大类:汽化(>600℃)、裂解(370~530℃,0.1~0.5 MPa)和液化(230~330℃,5~20 MPa)[13]。Karagöze和Yuan等[14-15]研究了生物质的低温水热液化的可能性及对硫酸盐木质素液体油得率的影响,发现在反应温度为180℃时,生物油的得率随反应时间的延长而增加,与此同时,加入Ca(OH)2有助于提高液化产物的得率。Pińkowska等[16]探讨了在亚临界和超临界水条件下温度对碱木质素降解的影响,发现温度的升高促进了木质素的解聚和再聚合。研究者不仅对水热条件下液体产物有所研究,同时,对生成的固体焦炭也进行了探索。Sasaki等[17]研究了碱木质素在近临界和超临界水中得到的主要液体产物为单体化合物,也发现了在降解过程中,焦炭功能基的分布发生了很大变化;Hu等[18]研究了黑液中木质素在280~365℃反应下焦炭的物理形态、得率、热稳定性和功能基的变化,发现在330℃条件下获得的焦炭具有最大的比表面积和孔隙度,同时X射线检测表明较高温度条件下制备的焦炭具有较高的结晶度。Yang等[19]研究了玉米芯木质素的水热降解,分析了温度和时间对五大降解产物(气体、挥发性有机化合物、水溶性油、重油和固体残渣)的影响,结果表明,得到的重油可以用于酚醛树脂的合成,但水热降解得到的液体产物种类较多,不易分离提纯及后续利用,因此存在一定局限。
除水热体系降解外,一些学者采用有机溶剂对木质素进行降解,研究较多的为醇类,醇溶剂的使用具有以下优点:1)醇的存在可促进木质素降解为可回收的液体燃料;2)木质素和低分子量的木质素产物可以较好地溶解在醇-水介质中,避免再聚合;3)低沸点醇易于后续分离和回收利用。因此,醇-水共混溶剂得到了广泛关注。Ye等[20]对乙醇-水混合溶液中水热解聚棉杆木质素进行了研究,发现所得产物主要以杂环和酚型结构为主,且反应条件(时间、温度及乙醇浓度)对液体产物均有较大影响;Cheng等[21]先后研究了生物质在亚/超临界醇和醇-水混合溶剂中的高效液化降解,认为50%的醇水混合溶液可以使生物质有效液化,获得的生物油产物主要以酚型化合物及其衍生物为主,其次为醛、长链酮及醇和有机酸等化合物。Hu和Ni等[22-23]对毛竹生物质在乙醇溶剂中的反应进行了研究,得到木质素转化成酚的优化条件,同时避免了纤维素和半纤维素的转化,并解析了木质素侧链C—C键断裂的机理。除乙醇外,其他有机溶剂也被尝试应用。Saisu等[24]分析木质素在超临界水-苯酚混合溶剂中转化的结果表明,苯酚能够促进木质素分解为低分子片断,但苯酚可与降解产物反应,生成烷基苯酚。Yuan等[25]研究了稻草在乙醇-水和2-异丙醇-水溶液中的亚/超临界液化,结果表明当2-异丙醇与水溶液体积比为1∶1时,所获的生物油最大得率为39.7%。同时,Jiang等[26]利用水-四氢呋喃溶剂将玉米芯残渣中木质素高得率、高选择性地转化成了单酚类化合物。
近年来,研究发现离子液体具有较好的溶解纤维素能力,为生产各种新的化合物提供了平台[27]。同时,一些学者对木质素在离子液体中的降解过程及机理也进行了研究[28-29]。Cox等[30]分析了相同阳离子不同阴离子的离子液体对木质素模型物的降解,发现反应不仅与离子液体的酸性有关,同时也与阴离子的性质有关(图1)[30],而Hart等[31]就离子液体中阴、阳离子对木质素溶解性的影响进行了研究,发现阳离子的影响远大于阴离子。Zakzeski等[32]研究了不同提取木质素和木质素模型物在离子液体、过渡金属和分子氧存在下的氧化降解反应,对反应温度、氧压和NaOH的用量等因素均进行了探究,发现六水氯化钴在所用的离子液体中对氧化反应较为有效。除离子液体与水混合外,还有一些离子液体共混溶剂逐渐被用于生物质降解反应,如二甲亚砜[33],但离子液体成本高仍是限制其广泛使用的瓶颈问题之一。在3种体系中,水热体系由于其经济、环保的特点最具发展前景,但在处理过程中如何避免焦炭生成是需要解决的主要问题。
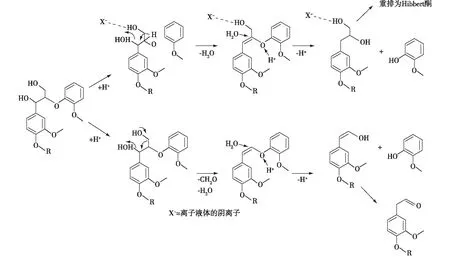
图1 离子液体中β-O-4′芳基醚的断裂机理Fig. 1 Mechanism of β-O-4′ aryl ether bond cleavage in ionic liquids
2 木质素化学降解常用催化剂
木质素催化转化为化学品是当前研究的热点领域之一。催化剂的存在可提高木质素的转化率,抑制木质素大分子缩合及焦炭的生成。一般情况下,催化剂会选择性地断裂木质素单元之间连接键,从而能够高选择性获得某一类特定化合物。普通的催化剂有液体酸,如H2SO4、HCl、H3PO4等,这些液体酸的催化活性较高,但大量使用消费较高,因为该过程会导致设备严重被腐蚀,且废酸的分离和中和成本高[34]。同时,碱催化剂(KOH和NaOH)在木质素降解过程中也表现出较好效果,该过程被称为碱性催化解聚[35-37]。近年来,诸多学者致力于开发用于生物质解聚的固体催化剂,如全氟磺酸、大孔树脂、磺酸化的无定形碳、介孔二氧化硅、H形沸石(HZSM-5)和杂多酸,甚至金属氧化物(如γ-Al2O3)[38-41],其中固体的Brønsted酸被认为是一种有效的催化剂[42]。有研究使用酸性金属盐作为催化剂,如金属氯化物[43],研究表明FeCl3、CuCl2和 AlCl3为断裂愈创木基(G)-愈创木基(G)单元间β-O-4′键的有效催化剂。在FeCl3和CuCl2共存时,温度为150℃、反应时间为120 min条件下 GG(愈创木基-愈创木基单元)的转化率为100%,约70%的β-O-4′键被水解转化为愈创木酚;而单独使用AlCl3做催化剂时,在GG结构中约80%的β-O-4′键被水解,同时也证实了AlCl3在断裂紫丁香基(V)-愈创木基(G)的β-O-4′键时比FeCl3和CuCl2更有效。在使用AlCl3作催化剂,反应温度为150℃、反应时间240 min时, VG(紫丁香基-愈创木基单元)约有75%的β-O-4′键被水解,表明该种酸性催化剂的催化活性与金属氯化物水解原位形成的HCl有关[44]。
除上述催化剂,还有各种重金属催化剂、赋予金属功能的离子液体和基于铁、钴、镍的复合催化剂等。近年来,自行合成的复合催化剂颇受关注,主要包括有机或无机的金属复合催化剂,如Ni/C,Cu/MgO-Al2O3、Ni-SIPr等[45-47]。Ye等[48]研究了酶解玉米秆木质素的温和水解(温度200~250℃),在使用Ru/C,Pt/C或 Pd/C作催化剂时,产物中主要组分为4-乙基苯酚(3.1%)和4-乙基愈创木酚(1.4%)。Toledano等[49]将金属纳米粒子镍(Ni)、钯(Pd)、铂(Pt)和钌(Ru)负载在Al-SBA-15上,在微波条件下对有机溶剂木质素进行了处理,结果表明,采用Ni或Pd作为催化剂时,主要产物为邻苯二甲酸二乙酯。Song等[50]研究了在常用的醇类溶剂中添加Ni/C催化剂降解木质素的效果,发现在该过程中Ni基催化剂可提高反应活性和选择性,在木质素转化率为50%时生成单体酚的选择性最高可达97%,同时,该催化剂能够通过磁性分离循环回收,并具有较好的循环回用能力。因此,复合型的金属催化剂,不仅可使反应在较温和的条件下进行,而且还能够提高催化剂的选择性、稳定性和效率,同时可回收再用,具有广阔的应用前景。
3 木质素催化还原降解
木质素的还原降解易于生成一些简单的芳香族化学品,因此氢解是目前从木质素中生产酚类较有前景的方法。与热裂解相比,氢解的净转化率及单体酚得率更高,同时生成的焦炭更少。氢解过程为利用高温加压的分子氢在适当催化剂存在条件下,对木质素进行解聚和加氢脱氧。Meier等[51]研究了气体氢环境下使用NiMo铝硅酸盐催化剂对木质素进行催化氢解,获得的液体油得率为65%,发现氢气压力对转化率有显著影响,随氢压从5 MPa升至14 MPa,轻质油的得率从20%增至57%,而酚类组分从7.0%增至12.3%,同时氢的存在还能够抑制焦炭的生成。Thring等[52]的研究结果表明,增加氢压不仅能提高净转化率,而且可显著降低残渣量,相似的结果也被Meier等[53]发现,当氢压从5 MPa增至14 MPa时,剩余残渣量从32.0%降为1.9%。
基于安全等方面考虑,也有研究者选用其他可替代的液体氢解试剂。目前已有一些文献报道利用甲酸作为氢解试剂进行木质素降解、氢解反应[54-55],不仅可减少木质素缩合,还能够促进木质素还原产物的生成。在一定反应条件下,甲酸加热后会完全分解为CO2和活性氢,与木质素甲氧基的氧结合可生成水(图2)[54]。
括号内为T值,调整后的R2为0.2240,F值为7.35,并在5%显著水平下通过检验。由结果可知资本产出弹性α=0.6454,劳动产出弹性为0.3546。因为二阶差分后,弱化了常数项,所以常数项并不显著。将α值代入(3)式,即可得全要素生产率的增长率,结果见表2。
因解聚和加氢脱氧同
时进行,所以该溶剂分解反应可以一步生成低氧含量的单体。除甲酸外,一些可提供还原氢的醇、四氢化萘等也被用于木质素氢解,既作为溶剂也作为供氢体。Davoudzadeh等[56]采用苯酚做溶剂,利用四氢化萘对木质素进行氢解,并与木质素的裂解进行对比,结果表明氢解后的液体得率提高显著。Kudsy等[57]也对四氢化萘在氢解过程中的作用进行了探讨,添加四氢化萘增加了酚类化合物的得率,但对气体产率没有显著影响。
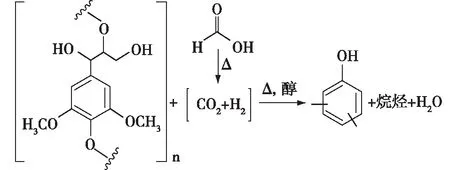
图2 甲酸存在时木质素的分解和脱甲氧基过程Fig. 2 Scheme of lignin decomposition and demethoxylation in the presence of formic acid
除还原剂外,在反应过程中添加不同的催化剂能够提高木质素氢解的程度,从而进一步提高目标产物的得率。近年来,研究者们已提出了各种利用均相、异相、有机或无机还原性催化剂促进氢解的反应。Sergeev等[58]阐述了一种用Ni(COD)2和N-杂环碳烯原位合成的催化剂高效实现木质素模型物选择性断裂连接键的方法。碱的加入可抑制芳环的氢解,该方法能够适用于大量的含取代基的二芳醚,对带有吸电子基的底物效果最好。用不带稳定配体的镍作前驱体,也可以实现富电子芳醚的有效断裂,同时产生镍的纳米粒子[59]。与二芳基底物相比,芳基烷基醚键裂解速率较低。使用氘示踪的烷基芳基醚的研究结果表明,底物断裂的机理为镍对芳环体系的配位引起C—O的嵌入和β-H消除,释放出甲醛,产生芳基-镍氢化物,最后经还原消除生成了脱氧的芳香族化合物(图3)[58-60]。
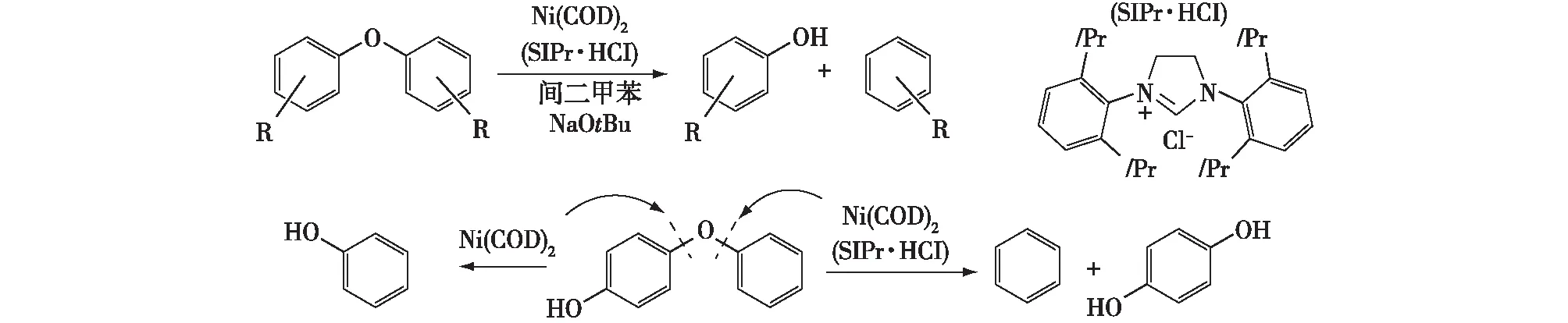
图3 镍基催化体系下木质素二芳醚模型物的裂解Fig. 3 Diaryl ether lignin model compounds cleaved by nickel catalyst systems
一种异相的Pd/C/Zn催化剂可导致木质素模型物中β-芳基醚键的断裂[61],在催化剂用量(质量分数)为5%、氢压为2 MPa 的甲醇溶剂中,木质素二聚体模型物β-O-4′醚键发生断裂,生成的丙基愈创木酚和愈创木酚得率为85%,还有少量醇的副产物;在上述条件下还探索了高聚合度木质素模型物的氢解,结果表明得到的丙基愈创木酚和愈创木酚为主要产物,得率分别为56%和44%(图4)[61]。

图4 使用Zn/Pd/C催化剂时β-芳基醚键的氢解断裂Fig. 4 β-aryl ether hydrogenolysis over a Zn/Pd/C catalyst
在各类还原性催化剂中,金属催化剂备受关注。单金属催化剂较早被研究应用,一般分为贵金属和非贵金属催化剂。贵金属通常负载在一些载体上,如活性炭、二氧化硅、氧化铝,以提高分散性。研究表明,在乙醇/水溶液中,温度为275℃,氢压为2 MPa,使用Ru/C、Pd/C和Pt/C催化剂反应1.5 h,可以从玉米秆和竹子木质素中高选择性地得到4-乙基酚类[62]。虽然贵金属催化剂效果较好,但价格昂贵,而且会引起苯环氢化,降低芳香化合物的得率。因此,非贵金属如Fe、Cu和Ni也被作为木质素氢解的催化剂。异相的FeMoP催化剂对于芳基醚和酚的氢解,特别是β-O-4′化合物具有较高选择性[63]。
近年来,研究发现添加第二种金属形成双金属催化剂有益于改进催化剂的几何和电子特性,两种金属的协同作用可以提高催化活性、选择性及稳定性。多种Ni基双金属催化剂能够在水中将β-O-4′模型物氢解成2-苯氧基-1-苯乙醇产物。与单组份Ni催化剂相比,NiAu、NiRu、NiRh和NiPd增强了催化效果,可显著提高催化活性及选择性。在测试的双金属催化剂中,NiAu的效果最好,但单金属Au催化剂在氢解中完全无效(图5)[64]。通过优化,Ni7Au3可使β-O-4′模型物在温和条件下(水相,130℃,氢压1 MPa,1 h)定量转化得到87%的单体得率。
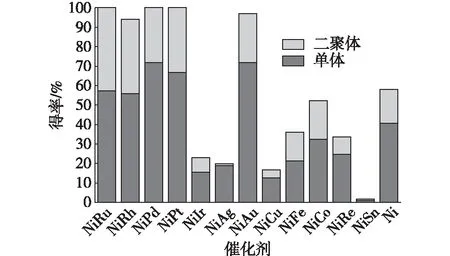
图5 使用纯Ni和双金属NiM催化剂的二聚体和单体得率Fig. 5 Dimer and monomer yields for pure Ni and bimetallic NiM catalysts
4 木质素催化氧化降解
木质素由于羟基、芳基醚键的存在能够发生氧化和氧化裂解,基于不同反应条件,木质素通过氧化降解反应可生成一些芳香醛、酮或羧酸类化合物,有益于木质素进一步工业化利用。目前,可利用的氧化剂有很多种,但由于过度氧化,要在保持较高转换价值的同时得到选择性高的产物仍面临巨大挑战,因此氧化型催化剂得到了广泛研究和应用。
最为常用的氧化剂是气体氧。此外,硝基苯、金属氧化物、过氧化氢等也是较受关注的氧化剂。Xiang等[62]研究了在水相介质中使用过氧化氢对木质素进行非催化氧化裂解。在碱性和酸性环境中,主要产物均包括单体和二羧酸,但所需反应条件不同。在碱性条件下木质素更易溶解,在温度为120℃、反应时间为5 min时,通过过氧化氢降解能使转化率达到98%;而在酸性条件下,温度为160℃、反应时间为10 min时,产物最高得率为97.4%。同时,在碱性环境下产物中较多乙二酸和甲酸;而在酸性环境下多产生甲酸和乙酸,但产物中香草醛、紫丁香醛或其他芳香醛、酸含量较少,这表明过氧化氢氧化作用较强。
相比较而言,硝基苯、金属氧化物和氧气是较温和的氧化剂,能保留木质素芳环并生产芳香醛。采用硝基苯作氧化剂的研究结果表明,使用硫酸盐木质素转化为香草醛的得率约为13%~14%[65-66]。硝基苯虽然是有效的氧化剂,但也为致癌物,因此,Masingale等[67]研究了用Cu2+、Fe3+或组合的金属有机结构作为氧化剂以代替木质素氧化中的硝基苯。此外,研究表明,使用分子氧作为氧化剂,可从水解的木质素、碱木质素和硫酸盐木质素中分别得到14.4%,8.0%和3.5%的醛结构[68],同时随氧压增加,水解木质素的催化氧化可得到更多醛结构。然而,研究发现在酸性条件下利用分子氧进行木质素氧化是不合理的。如Gonçalves等[69]在酸性条件下进行了有机溶剂木质素的催化氧化反应,结果表明醛的得率较低,因此目前研究较多的为木质素的碱性氧化,Tarabanko等[70]提出了木质素碱性氧化成香草醛的机理,如图6所示。
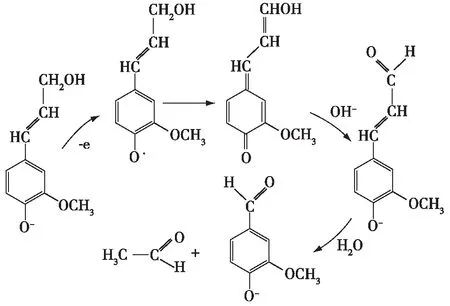
图6 木质素碱性氧化成香草醛的反应机理Fig. 6 Reaction mechanism for vanillin formation during alkaline oxidation of lignin
Badamali等[73]阐述了微波辐射条件下,以介孔MCM-41、HMS、SBA-15和无定形氧化硅作为催化剂,过氧化氢为氧化剂和乙腈为溶剂来氧化4-羟基-3-甲氧基苯乙醇的方法。结果表明,在30 min辐射后,该体系中生成了香草乙酮、香草醛和2-甲氧基苯醌,并发现其中无定形氧化硅活性很强,但缺乏选择性(图7)[73]。
Tonucci等[74]对不同光催化体系实现木质素脂肪族C—C键的选择性断裂并保留芳环进行了研究,结果表明,两种商业的钙基和氨基木质素衍生物在H2O2存在条件下主要生成了香草醛和松柏醛,同时,松柏醇、苯酚、2-羟基苯甲醇和水杨醛也被检测到。Hanson等[75]分析了钒基催化剂对木质素键的选择性断裂。在二醇模型体系中的研究表明,在有氧条件下能够实现键的裂解,同时其他模型物也可发生氧化断裂,且苯基取代基促进了反应[76]。Son等[77]报道了用催化剂[V1]可实现β-O-4′键有效断裂,同时其他钒催化剂也被发现可引起木质素模型物连接键选择性断裂[78]。采用13C示踪木质素二聚模型物发现,钒催化剂能够引发不同位置的氧化键裂[79]。钒催化剂[V2]打断了苄基的C—C键,生成了2,6-二甲氧基苯醌和相应的醛,相反,钒催化剂[V1]导致了C—O键的选择性断裂。此外,研究还发现使用催化剂[V2]打断苄基的C—C键仅限于酚型的底物(图8)[77-79]。
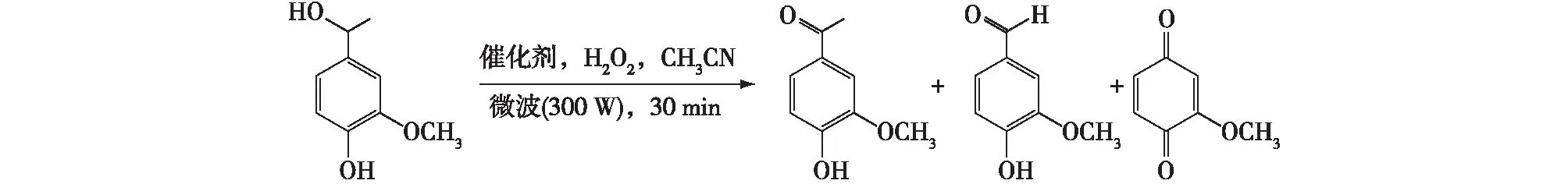
图7 介孔氧化硅催化氧化4-羟基-3-甲氧基苯乙醇Fig. 7 Oxidation of apocynol over mesoporous silica catalysts
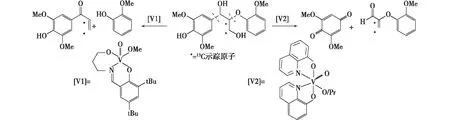
图8 两种不同的钒基催化剂对β-O-4′木质素模型物的选择性氧化断裂Fig. 8 Selective oxidative cleavage of a β-O-4′ lignin model using two different vanadium based catalysts
5 展 望
木质素是由苯丙烷基以C—O或C—C键结合形成的三维网状结构,富含芳环的结构特性使得木质素成为制备芳香类化学品的重要原料,基于化学催化手段将其高效转化为高值产品具有广泛研究前景。尽管有关木质素催化降解方面的研究已取得了显著进展,但该领域研究尚存在诸多未解决的问题,学者探索的多种木质素催化降解催化剂和反应体系,普遍存在反应条件苛刻(高温和高压)、反应转化率低、目标产物选择性差等缺点;在溶剂选择方面,水热体系由于其经济、环保的特点最具发展前景,但在处理过程中如何避免焦炭生成是急需解决的问题;而催化剂仍以过渡金属及贵金属为主,价格昂贵且容易失活。因此,如何开发高效低成本催化剂,利用温和反应体系,在保证产物得率和选择性的同时,不损失催化剂的稳定性和活性,是木质素大分子高效转化与利用所面临的较大挑战。
[ 1 ]GUO D J, CHEN F, KENTARO I. Downregulation of caffeic acid 3-O-methyltransferase and caffeoyl CoA 3-O-methyltransferase in transgenic alfalfa:impacts on lignin structure and implications for the biosynthesis of G and S lignin[J]. Plant Cell, 2001, 13:73-88.
[ 2 ]WANG H, BLOCK L E, ROGERS R D. Catalytic conversion of biomass in ionic liquids[J]. RSC Catalysis, 2014(15):1-19.
[ 3 ]BOERJAN W, RALPH J, BAUCHER M. Lignin biosynthesis[J]. Annual Review of Plant Physiology, 2003, 54:519-546.
[ 4 ]ZHONG R Q, MORRISON W H, HIMMELSBACH D S. Essential role of caffeoyl coenzyme a O-methyltransferase in lignin biosynthesis in woody poplar plants[J]. Plant Physiol, 2000, 124:563-577.
[ 5 ]SAIDI M, SAMIMI F, KARIMIPOURFARD D, et al. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation[J]. Energy & Environmental Science, 2014, 7(1):103-129.
[ 6 ]王翔, 蒋帅南, 陈敏智, 等. 木质素基碳纤维研究进展[J]. 林业工程学报, 2016, 1(1):83-87. WANG X, JIANG S N, CHEN M Z, et al. Review of research progress on lignin-based carbon fibers[J]. Journal of Forestry Engineering, 2016, 1(1):83-87.
[ 7 ]RAGAUSKAS A J, BECKHAM G T, BIDDY M J, et al. Lignin valorization:improving lignin processing in the biorefinery[J]. Science, 2014, 344(6185):1246843.
[ 8 ]ZHANG L, YOU T, TIAN Z, et al. Interconnected hierarchical porous carbon from lignin-derived byproducts of bioethanol production for ultra-high performance supercapacitors[J]. ACS Applied Materials & Interfaces, 2016, 8(22):13918-13925.
[ 9 ]PANDEY M P, KIM C S. Lignin depolymerization and conversion:a review of thermochemical methods[J]. Chemical Engineering & Technology, 2011, 34(1):29-41.
[10]CHEN F, DIXON R A. Lignin modification improves fermentable sugar yields for biofuel production[J]. Nature Biotechnology, 2007, 25:759-761.
[11]FU C, MIELENZ J R, XIAO X. Genetic manipulation of lignin reduces recalcitrance and improves ethanol production from switchgrass[J]. Proceedings of the National Academy of Sciences, 2011, 108:3803-3808.
[12]CHATEL G, ROGERS R D.Review:oxidation of lignin using ionic liquids:an innovative strategy to produce renewable chemicals[J]. ACS Sustainable Chemistry and Engineering, 2013, 2(3):322-339.
[13]ZHOU X F. Conversion of kraft lignin under hydrothermal conditions[J]. Bioresource Technology, 2014, 170:583-586.
[14]KARAGÖZ S, BHASKAR T, MUTO A, et al. Low-temperature hydrothermal treatment of biomass:effect of reaction parameters on products and boiling point distributions[J]. Energy & Fuels, 2004, 18(1):234-241.
[15]YUAN Z, CHENG S, LEITCH M, et al. Hydrolytic degradation of alkaline lignin in hot-compressed water and ethanol[J]. Bioresource Technology, 2010, 101(23):9308-9313.
[17]SASAKI W M, GOTO M.Recovery of phenolic compounds through the decomposition of lignin in near and supercritical water[J]. Chemical Engineering and Processing:Process Intensification, 2008, 47(9):1609-1619.
[18]HU J, SHEN D, WU S,et al. Effect of temperature on structure evolution in char from hydrothermal degradation of lignin[J]. Journal of Analytical and Applied Pyrolysis, 2014, 106:118-124.
[19]YANG S, YUAN T Q, LI M F, et al. Hydrothermal degradation of lignin:products analysis for phenol formaldehyde adhesive synthesis[J]. International Journal of Biological Macromolecules, 2015, 72:54-62.
[20]YE Y, FAN J, CHANG J. Effect of reaction conditions on hydrothermal degradation of cornstalk lignin[J]. Journal of Analytical and Applied Pyrolysis, 2012, 94:190-195.
[21]CHENG S, D’CRUZ I, WANG M, et al. Highly efficient liquefaction of woody biomass in hot-compressed alcohol-water co-solvents[J]. Energy & Fuels, 2010, 24(9):4659-4667.
[22]HU L, LUO Y, CAI B,et al. The degradation of the lignin inPhyllostachysheterocyclacv.pubescensin an ethanol solvothermal system[J]. Green Chemistry, 2014, 16(6):3107-3116.
[23]NI Y, HU Q. Alcell lignin solubility in ethanol-water mixtures[J]. Journal of Applied Polymer Science, 1995, 57(12):1441-1446.
[24]SAISU M, SATO T, WATANABE M,et al. Conversion of lignin with supercritical water-phenol mixtures[J]. Energy & Fuels, 2003, 17(4):922-928.
[25]YUAN X Z, LI H, ZENG G M,et al. Sub-and supercritical liquefaction of rice straw in the presence of ethanol-water and 2-propanol-water mixture[J]. Energy, 2007, 32(11):2081-2088.
[26]JIANG Z, HE T, LI J,et al. Selective conversion of lignin in corncob residue to monophenols with high yield and selectivity[J]. Green Chemistry, 2014, 16(9):4257-4265.
[27]YU H M, HU J, FAN J, et al. One-pot conversion of sugars and lignin in ionic liquid and recycling of ionic liquid[J]. Industrial & Engineering Chemistry Research, 2012, 51:3452-3457.
[28]COX B J, EKERDT J G. Depolymerization of oak wood lignin under mild conditions using the acidic ionic liquid 1-H-3-methylimidazolium chloride as both solvent and catalyst[J]. Bioresource Technology, 2012, 118:584-588.
[29]DAI J, PATTI A F, SAITO K. Recent developments in chemical degradation of lignin:catalytic oxidation and ionic liquids[J]. Tetrahedron letters, 2016,57(45):4945-4951.
[30]COX B J, JIA S, ZHANG Z C,et al. Catalytic degradation of lignin model compounds in acidic imidazolium based ionic liquids:Hammett acidity and anion effects[J]. Polymer Degradation and Stability, 2011, 96(4):426-431.
[31]HART W E, HARPER J B, ALDOUS L. The effect of changing the components of an ionic liquid upon the solubility of lignin[J]. Green Chemistry, 2015, 17(1):214-218.
[32]ZAKZESKI J, JONGERIUS A L, WECKHUYSEN B M.Transition metal catalyzed oxidation of Alcell lignin, soda lignin, and lignin model compounds in ionic liquids[J]. Green Chemistry, 2010, 12(7):1225-1236.
[33]ANDANSON J M, BORDES E, DEVÉMY J, et al. Understanding the role of co-solvents in the dissolution of cellulose in ionic liquids[J]. Green Chemistry, 2014, 16(5):2528-2538.
[34]ZHOU C H, XIA X, LIN C X, et al. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels[J]. Chemical Society Reviews, 2011, 40(11):5588-5617.
[35]MILLER J E, EVANS L, LITTLEWOLF A, et al. Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents[J]. Fuel, 1999, 78(11):1363-1366.
[36]WATANABE M, INOMATA H, OSADA M, et al. Catalytic effects of NaOH and ZrO2for partial oxidative gasification of n-hexadecane and lignin in supercritical water[J]. Fuel, 2003, 82(5):545-552.
[37]NENKOVA S, VASILEVA T, STANULOV K. Production of phenol compounds by alkaline treatment of technical hydrolysis lignin and wood biomass[J]. Chemistry of Natural Compounds, 2008, 44(2):182-185.
[38]VYVER S V D, PENG L, GEBOERS J, et al. Sulfonated silica/carbon nanocomposites as novel catalysts for hydrolysis of cellulose to glucose[J]. Green Chemistry, 2010, 12(9):1560-1563.
[39]HEGNER J, PEREIRA K C, DEBOEF B, et al. Conversion of cellulose to glucose and levulinic acid via solid-supported acid catalysis[J]. Tetrahedron Letters, 2010, 51(17):2356-2358.
[40]SUGANUMA S, NAKAJIMA K, KITANO M, et al. Hydrolysis of cellulose by amorphous carbon bearing SO3H, COOH, and OH groups[J]. Journal of the American Chemical Society, 2008, 130(38):12787-12793.
[41]TIAN J, WANG J, ZHAO S, et al. Hydrolysis of cellulose by the heteropoly acid H3PW12O40[J]. Cellulose, 2010, 17(3):587-594.
[42]DHEPE P L, OHASHI M, INAGAKI S, et al. Hydrolysis of sugars catalyzed by water-tolerant sulfonated mesoporous silicas[J]. Catalysis Letters, 2005, 102(3-4):163-169.
[43]WU M, ZHAO D H, PANG J H, et al. Separation and characterization of lignin obtained by catalytic hydrothermal pretreatment of cotton stalk[J]. Industrial Crops and Products, 2015, 66:123-130.
[44]JIA S, COX B J, GUO X, et al. Hydrolytic cleavage ofβ-O-4 ether bonds of lignin model compounds in an ionic liquid with metal chlorides[J]. Industrial & Engineering Chemistry Research, 2010, 50(2):849-855.
[45]SONG Q, CAI J, ZHANG J, et al. Hydrogenation and cleavage of the CO bonds in the lignin model compound phenethyl phenyl ether over a nickel-based catalyst[J]. Chinese Journal of Catalysis, 2013, 34(4):651-658.
[46]STRASSBERGER Z, ALBERTS A H, LOUWERSE M J, et al. Catalytic cleavage of ligninβ-O-4 link mimics using copper on alumina and magnesia-alumina[J]. Green Chemistry, 2013, 15(3):768-774.
[47]SAWATLON B, WITITSUWANNAKUL T, TANTIRUNGROTECHAI Y, et al. Mechanism of Ni N-heterocyclic carbene catalyst for C—O bond hydrogenolysis of diphenyl ether:a density functional study[J]. Dalton Transactions, 2014, 43(48):18123-18133.
[48]YE Y, ZHANG Y, FAN J, et al. Selective production of 4-ethylphenolics from lignin via mild hydrogenolysis[J]. Bioresource Technology, 2012, 118:648-651.
[49]TOLEDANO A, SERRANO L, PINEDA A, et al. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis:catalyst screening[J]. Applied Catalysis B:Environmental, 2014, 145:43-55.
[50]SONG Q, WANG F, CAI J, et al. Lignin depolymerization(LDP) in alcohol over nickel-based catalysts via a fragmentation-hydrogenolysis process[J]. Energy & Environmental Science, 2013, 6(3):994-1007.
[51]MEIER D, ANTE R, FAIX O. Catalytic hydropyrolysis of lignin:influence of reaction conditions on the formation and composition of liquid products[J]. Bioresource Technology, 1992, 40(2):171-177.
[52]THRING R W, BREAU J. Hydrocracking of solvolysis lignin in a batch reactor[J]. Fuel, 1996, 75(7):795-800.
[53]MEIER D, BERNS J, GRÜNWALD C, et al. Analytical pyrolysis and semicontinuous catalytic hydropyrolysis of organocell lignin[J]. Journal of Analytical and Applied Pyrolysis, 1993, 25:335-347.
[54]XU W, MILLER S J, AGRAWAL P K, et al. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid[J]. Chemsuschem, 2012, 5(4):667-675.
[55]TOLEDANO A, SERRANO L, BALU A M, et al. Fractionation of organosolv lignin from olive tree clippings and its valorization to simple phenolic compounds[J]. Chemsuschem, 2013, 6(3):529-536.
[56]DAVOUDZADEH F, SMITH B, AVNI E, et al. Depolymerization of lignin at low pressure using Lewis acid catalysts and under high pressure using hydrogen donor solvents[J]. Holzforschung, 1985, 39(3):159-166.
[57]KUDSY M, KUMAZAWA H, SADA E. Pyrolysis of kraft lignin in molten ZnCl2-KCl media with tetralin vapor addition[J]. The Canadian Journal of Chemical Engineering, 1995, 73(3):411-415.
[58]SERGEEV A G, HARTWIG J F. Selective, nickel-catalyzed hydrogenolysis of aryl ethers[J]. Science, 2011, 332:439-443.
[59]SERGEEV A G, WEBB J D, HARTWIG J F. A heterogeneous nickel catalyst for the hydrogenolysis of aryl ethers without arene hydrogenation[J]. Journal of the American Chemical Society, 2012, 134(50):20226-20229.
[60]KELLEY P, LIN S, EDOUARD G, et al. Nickel-mediated hydrogenolysis of C—O bonds of aryl ethers:what is the source of the hydrogen?[J]. Journal of the American Chemical Society, 2012, 134(12):5480-5483.
[61]PARSELL T H, OWEN B C, KLEIN I, et al.Cleavage and hydrodeoxygenation(HDO) of C-O bonds relevant to lignin conversion using Pd/Zn synergistic catalysis[J]. Chemical Science, 2013, 4(2):806-813.
[62]XIANG Q, LEE Y Y. Oxidative cracking of precipitated hardwood lignin by hydrogen peroxide[J]. Applied Biochemistry and Biotechnology, 2000, 84(1-9):153-162.
[63]RENSEL D J, ROUVIMOV S, GIN M E, et al. Highly selective bimetallic FeMoP catalyst for C-O bond cleavage of aryl ethers[J]. Journal of Catalysis, 2013, 305:256-263.
[64]ZHANG J, ASAKURA H, VAN RIJN J, et al. Highly efficient, NiAu-catalyzed hydrogenolysis of lignin into phenolic chemicals[J]. Green Chemistry, 2014, 16(5):2432-2437.
[65]MATHIAS A L, RODRIGUES A E. Production of vanillin by oxidation of pine kraft lignins with oxygen[J]. Holzforschung, 1995, 49(3):273-278.
[66]VILLAR J C, CAPEROS A, GARCA-OCHOA F. Oxidation of hardwood kraft-lignin to phenolic derivatives. Nitrobenzene and copper oxide as oxidants[J]. Journal of Wood Chemistry and Technology, 1997, 17(3):259-285.
[67]MASINGALE M P, ALVES E F, BOSE S K, et al. An oxidant to replace nitrobenzene in lignin analysis[J]. Bioresources, 2009, 4(3):1139-1146.
[68]XIANG Q, LEE Y Y. Production of oxychemicals from precipitated hardwood lignin[J]. Applied Biochemistry and Biotechnology, 2001, 91:71-80.
[69]GONÇALVES A R, SCHUCHARDT U. Oxidation of organosolv lignins in acetic acid [C].∥ Twentieth Symposium on Biotechnology for Fuels and Chemicals. Clifton, New Jersey: Humana Press, 1999:127-132.
[70]TARABANKO V E, PETUKHOV D V, SELYUTIN G E. New mechanism for the catalytic oxidation of lignin to vanillin[J]. Kinetics and Catalysis, 2004, 45(4):569-577.
[71]PAN K, TIAN M, JIANG Z H, et al. Electrochemical oxidation of lignin at lead dioxide nanoparticles photoelectrodeposited on TiO2nanotube arrays[J]. Electrochimica Acta, 2012, 60:147-153.
[72]SHIRAISHI T, TAKANO T, KAMITAKAHARA H, et al. Studies on electrooxidation of lignin and lignin model compounds. Part 1:direct electrooxidation of non-phenolic lignin model compounds[J]. Holzforschung, 2012, 66(3):303-309.
[73]BADAMALI S K, LUQUE R, CLARK J H, et al. Unprecedented oxidative properties of mesoporous silica materials:towards microwave-assisted oxidation of lignin model compounds[J]. Catalysis Communications, 2013, 31:1-4.
[74]TONUCCI L, COCCIA F, BRESSAN M, et al. Mild photocatalysed and catalysed green oxidation of lignin:a useful pathway to low-molecular-weight derivatives[J]. Waste and Biomass Valorization, 2012, 3(2):165-174.
[75]HANSON S K, BAKER R T, GORDON J C, et al. Aerobiccxidation of pinacol by vanadium(V) dipicolinate complexes:evidence for reduction to vanadium(III)[J]. Journal of the American Chemical Society, 2008, 131(2):428-429.
[76]HANSON S K, BAKER R T, GORDON J C, et al. Aerobic oxidation of lignin models using a base metal vanadium catalyst[J]. Inorganic Chemistry, 2010, 49(12):5611-5618.
[77]SON S, TOSTE F D. Non-Oxidative vanadium-catalyzed C—O bond cleavage:application to degradation of lignin model compounds[J]. Angewandte Chemie International Edition, 2010, 49(22):3791-3794.
[78]ZHANG G, SCOTT B L, WU R, et al. Aerobic oxidation reactions catalyzed by vanadium complexes of bis(phenolate) ligands[J]. Inorganic Chemistry, 2012, 51(13):7354-7361.
[79]HANSON S K, WU R, SILKS L A. C-C or C-O bond cleavage in a phenolic lignin model compound:selectivity depends on vanadium catalyst[J]. Angewandte Chemie International Edition, 2012, 51(14):3410-3413.
Recent development on conversion of lignin intoaromatics based on catalytical depolymerization
ZHANG Xueming, WU Miao, XU Feng*
(CollegeofMaterialsScienceandTechnology,BeijingForestryUniversity,Beijing100083,China)
Lignin is one of the three main components of lignocellulose and the most abundant natural aromatic polymer on the earth. Potentially lignin can be converted into high-value aromatic chemicals to replace the nonrenewable fossil resources by catalytical degradation. This research area has become one of the hottest topics in the related field. In this review, recent developments regarding the catalytical degradation of lignin are summarized. The types of reducing agents, oxidizing agents used in the catalytic reduction and oxidation reaction of lignin were discussed based on the mechanisms of catalytical reaction. The mechanism and aromatic products of these reaction systems were also summarized. Moreover, common solvents and catalysts used in lignin chemical degradation were discussed in detail. Finally, future outlook regarding the conversion of lignin into high-value chemicals were discussed. The development and synthesis of highly efficient and selective as well as cost efficient catalysts are the key points to break the bottleneck of this research field.
lignin; degradation; catalyst; reduction; oxidation
2016-11-28
2017-01-14
国家杰出青年科学基金(31225005);国家自然科学基金(31470606)。
张学铭,男,教授,研究方向为天然产物改性及有机合成。通信作者:许凤,女,教授。E-mail:xfx315@bjfu.edu.cn
TQ35
A
2096-1359(2017)04-0001-09
猜你喜欢
小学阅读指南·低年级版(2022年5期)2022-05-09
当代水产(2021年10期)2022-01-12
上海包装(2019年8期)2019-11-11
第一财经(2019年8期)2019-08-26
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
天津造纸(2016年1期)2017-01-15
中国造纸学报(2015年1期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
