
多种有序钙钛矿结构的高压制备与特殊物性∗
2017-07-31 05:59殷云宇王潇邓宏芟周龙戴建洪龙有文2
物理学报 2017年3期
殷云宇王潇邓宏芟周龙戴建洪龙有文2)†
1)(中国科学院物理研究所,北京凝聚态物理国家实验室(筹),北京 100190)2)(量子物质科学协同创新中心,北京 100190)(2017年1月17日收到;2017年1月18日收到修改稿)
多种有序钙钛矿结构的高压制备与特殊物性∗
殷云宇1)王潇1)邓宏芟1)周龙1)戴建洪1)龙有文1)2)†
1)(中国科学院物理研究所,北京凝聚态物理国家实验室(筹),北京 100190)2)(量子物质科学协同创新中心,北京 100190)(2017年1月17日收到;2017年1月18日收到修改稿)
具有ABO3钙钛矿或类似结构的强关联电子体系是凝聚态物理研究的重要前沿领域,而高压是制备新型钙钛矿特别是A位与/或B位有序钙钛矿材料的有效手段.在这些有序钙钛矿中,因A,B位可同时容纳过渡金属离子,因而可导致A-A,B-B,A-B等多种磁电相互作用的出现,进而诱导系列新颖有趣的物理现象.本文介绍高压下制备的几种化学式为的新型A位有序钙钛矿以及化学式为的A,B位同时有序的钙钛矿体系.在LaMn3Cr4O12中发现了具有立方钙钛矿结构的磁电多铁性,为多铁新材料探索与新机理研究提供范例;在CaCu3Fe2Os2O12中发现了远高于室温的亚铁磁半导体行为,并指出A位磁性离子的引入可大大增加磁相互作用强度从而大幅度提高磁有序温度;在LaMn3Ni2Mn2O12中观察到A位磁性离子调控的B位Ni2+/Mn4+子晶格正交自旋有序结构.以上研究结果为探索新型磁电多功能钙钛矿材料提供了重要参考.
高压合成,有序钙钛矿,多铁性,自旋有序
1 引 言
在ABO3钙钛矿中(图1(a)),A位往往由碱土金属、碱金属或稀土离子占据,形成AO12配位多面体,主要起到支撑结构的作用;而B位通常由过渡金属(transition metal,TM)离子(包括3d,4d,5d等)占据,形成BO6配位的八面体,很大程度上决定了材料的电子性质.由于A,B离子间可形成不同的电荷组态(包括A1+B5+,A2+B4+,A3+B3+,A4+B2+等不同类型),钙钛矿不仅具有灵活多变的晶体构型,同时也展示了丰富多彩的物理性质与功能特性,譬如压电、铁电、高温超导、巨磁电阻、多铁性等[1−19].在凝聚态物理研究的近几十年中,钙钛矿成为强关联电子体系的重要研究对象,众多新颖有趣的量子衍生现象在钙钛矿中不断被发现[20−25].
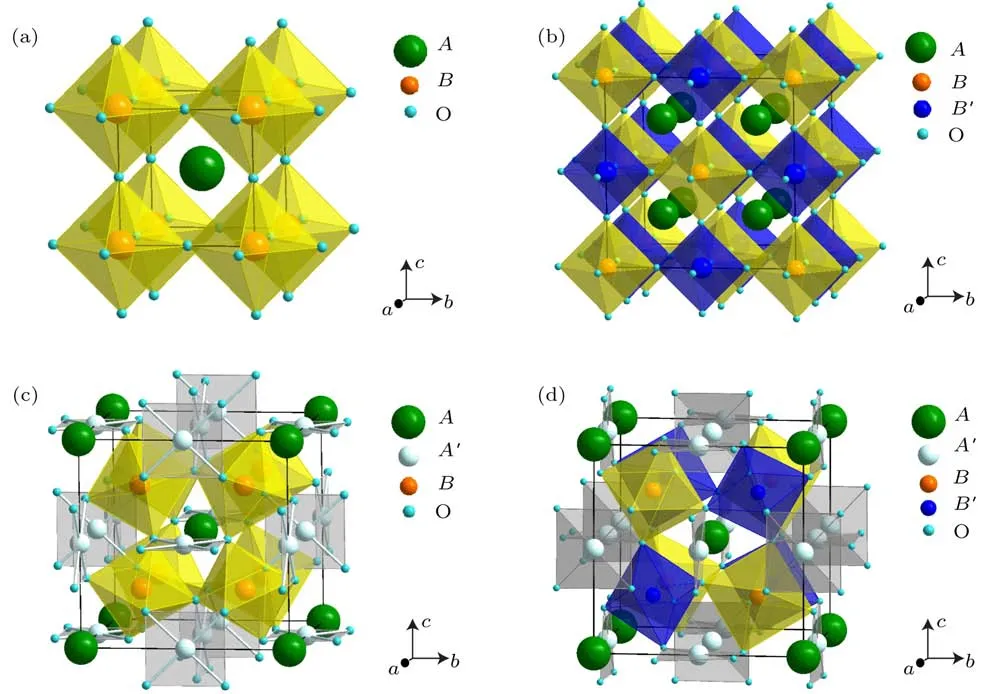
图1 不同类型钙钛矿晶体结构示意图 (a)简单ABO3钙钛矿晶体结构;(b)B位有序双钙钛矿A2BB′O6晶体结构;(c)A位有序钙钛矿晶体结构;(d)A,B位同时有序四重钙钛矿晶体结构Fig.1.Schematic viewofd iff erent typesofperovskites.The crystalstructuresof(a)simple ABO3perovskite,(b)B-site ordered doub le perovskite A2BB′O6,(c)A-site ordered perovskiteand(d)both A-and B-site ordered quadruple perovskite.
正因为钙钛矿具有灵活多变的晶体结构,通过合适的元素替代,可以形成不同类型的有序结构.比如在简单ABO3钙钛矿中,用另外一种TM离子B′替代二分之一的B位离子,可形成化学式为A2BB′O6的B位有序双钙钛矿(图1(b)).最近广受关注的高温铁磁(ferromagnetic,FM)半金属Sr2FeMoO6以及具有更高居里温度(约725 K)的Sr2CrOsO6均是B位有序双钙钛矿的典型代表[26,27].TM离子除了占据钙钛矿的B位外,能否通过特殊的合成方法也引入到A位呢?研究表明,如果将四分之三的A位离子用特殊的TM离子A′替代,可以形成化学式为的A位有序钙钛矿(图1(c)).然而,由于A′位TM离子的半径大大低于传统A位离子半径,为了维持钙钛矿结构,BO6八面体之间会发生严重倾斜,以致B—O—B键角往往在140◦左右.虽然单个的BO6八面体可能是刚性的,但这种严重倾斜的有序钙钛矿通常只有在高压高温条件下才能合成.另一方面,虽然这种特殊有序钙钛矿的A位仍然形成AO12配位,但A′位形成平行四边形配位的A′O4单元,意味着具有强Jahn-Teller畸变的离子如Cu2+,Mn3+等更有利于占据A′位.因为A位有序钙钛矿的A′位与B位同时容纳TM,因此除了常见的B-B相互作用外,也存在新型的A′-A′相互作用以及A′-B位间的相互作用.这些相互作用的出现,势必导致新颖物理性质的出现.比如,在B位非磁的CaCu3Ge4O12与CaCu3Sn4O12中,A′位磁性Cu2+离子的相互作用可诱导长程FM有序[28];CaCu3Ti4O12在宽温区内展示了超高的且几乎恒定的介电常数[29];在R Cu3Fe4O12(R=La—Gd,Bi)等A位有序钙钛矿中发现了Cu-Fe金属间电荷转移与负热膨胀等功能性质[30−34].
2 A位有序立方钙钛矿磁电多铁性材料LaMn3Cr4O12
磁电多铁性材料是指同时具有长程磁有序与电极化有序,甚至这两种有序相互耦合的材料体系.钙钛矿是多铁性研究最重要的材料体系之一,例如多铁研究的明星材料BiFeO3和TbMnO3便具有钙钛矿结构[35,36].众所周知,立方钙钛矿晶格具有反演对称中心,结构对称性上不支持铁电性的出现.但是,磁电多铁材料电极化的产生除了晶体结构的因素外,也可以来自于特殊的自旋结构[37−40].原则上,如果立方钙钛矿的磁结构打破空间反演对称性,那么同样可以产生电极化.然而,这种现象在以往所有的立方钙钛矿中未曾发现.在A位有序钙钛矿中,TM离子同时占据A′位和B位,从而可引入多重磁电相互作用.因此,通过选取合适的A′,B位磁性离子,一方面可维持立方钙钛矿晶体结构,另一方面也可能形成特殊的自旋有序结构打破空间反演对称性,从而诱导磁电耦合多铁现象.实验表明,我们在高压高温条件下制得的LaMn3Cr4O12(LMCO)是第一个被发现的具有立方钙钛矿结构的磁电多铁性材料[41].
利用高温高压实验条件(8 GPa,1400 K),我们获得了高质量LMCO多晶样品[42].图2给出了该化合物在不同温度下的粉末X射线衍射(X-ray di ff raction,XRD)谱.结果表明在293—23 K温度范围内LMCO始终保持A位有序钙钛矿结构,具有立方的Im-3空间群,晶体结构示意图见图1(c).通过磁化率、磁化强度和比热测试,我们发现LMCO存在两个反铁磁(antiferromagnetic,AFM)相变,其相变温度分别为TMn~50 K和TCr~ 150 K(图3).
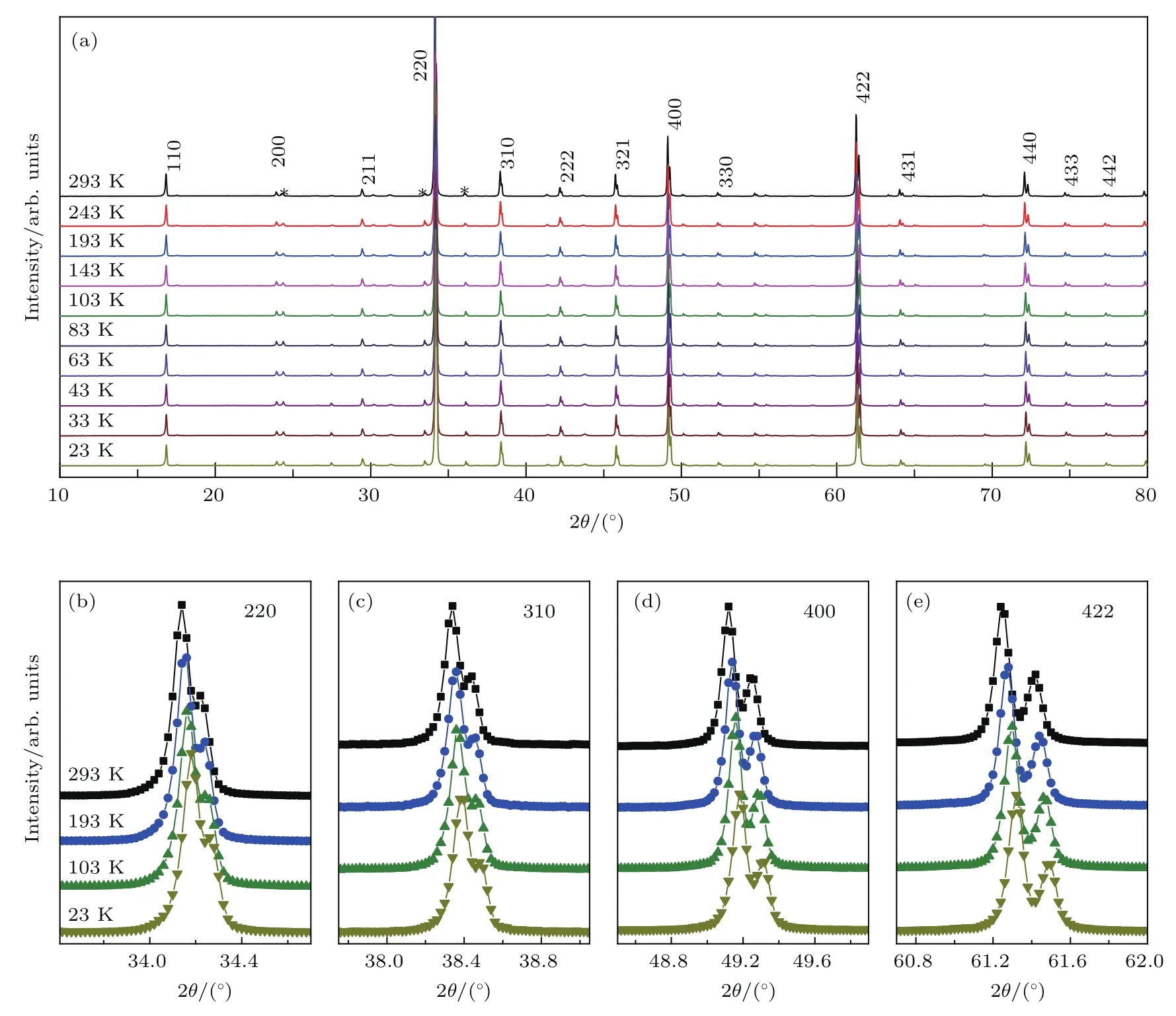
图2 (a)LMCO变温XRD,衍射峰根据立方Im-3空间群进行指标化,星号表示样品中少量的Cr2O3杂质;(b)—(e)几个主要衍射峰的放大图,清楚表明样品在293—23 K没有结构相变发生Fig.2.(a)Temperatu re dependent XRD of LMCO.The d i ff raction peaks are indexed based on the space groupof Im-3.The stars showthe d i ff raction peaks originating froma small amount of Cr2O3impu rity.(b)–(e)The en larged views for several representative di ff ractions peaks.Nostructu ral phase transition occu rs as temperature decreases from293 to23 K.
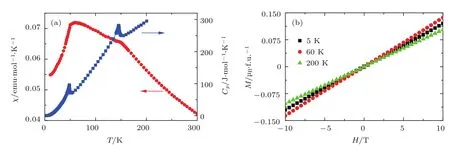
图3 (a)LMCO的磁化率和比热随温度的变化;(b)不同温度下LMCO的磁化强度随磁场的变化,其线性行为与中子粉末衍射揭示的G-型AFM有序一致Fig.3.(a)Temperature dependence of magnetic susceptibility and speci fi c heat of LMCO;(b)magnetization measured at various temperatu res,the linear magnetization behaviors are consistent with the collinear G-type AFMstructu re as revealed by neu tron powder d i ff raction.
为了探究LMCO的AFM相变起源,我们对该体系进行了中子粉末衍射(neutron powder di ff raction,NPD)实验(图4).与低温XRD结果一致,低温NPD表明,即使温度降低至2 K,LMCO仍然保持空间群为Im-3的A位有序立方钙钛矿晶体结构.同时,根据衍射峰(111)和(100)随温度的变化情况(图5),我们可以得知TCr~150 K的AFM相变来自B位Cr3+磁性离子子晶格的自旋有序,而TMn~50 K的AFM相变则源于A′位Mn3+磁性离子子晶格的自旋有序.进一步分析表明,不论是B位Cr3+子晶格还A′位Mn3+子晶格,都各自形成G型AFM结构,磁矩最可能均沿[111]方向平行排列(图6).显然,这种共线形式的AFM有序与实验所测得的线性磁化行为是一致的(图3(b)).

图4 不同温度下采集的LMCO的NPD数据及其精修结果,可观察到少量C r2O3和MnC r2O4杂质(<5 wt%)Fig.4.The NPD data as well as the Rietveld re fi nements for LMCOcollected at d i ff erent temperatu res.Asmall amount of Cr2O3and MnCr2O4impurity phases(<5 wt%)is observed.

图5 LMCO的(111)和(100)NPD峰的积分强度随温度的变化Fig.5. Temperatu re dependence of the integrated NPD intensities of(111)and(100)peaks of LMCO.
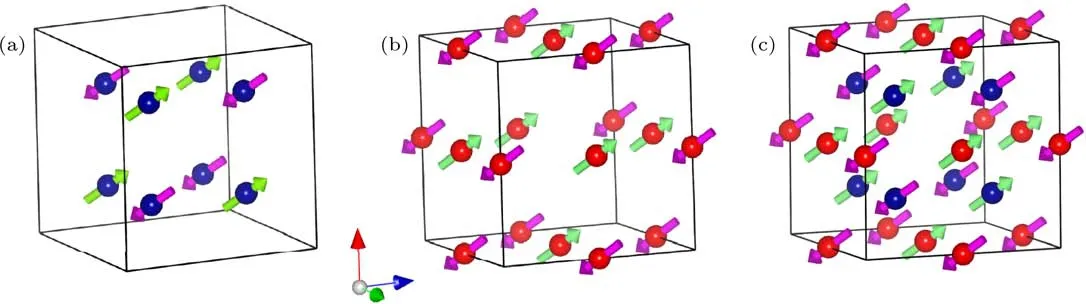
图6 (a),(b)分别给出了B位Cr子晶格和A′位Mn子晶格沿[111]方向的G型AFM结构;(c)Cr,Mn子晶格在TMn以下形成的总的磁结构;为了清楚起见,图中略去了La和O原子,其中,蓝色球代表C r原子,红色球代表Mn原子Fig.6.(a),(b)G-type AFMstructu re of the B-site Cr-sub lattice and the A′-site Mn-sub lattice with spin orientation along[111]d irection,respectively;(c)a complete set of spin alignment composed of Cr and Mn spins belowTMn.For clarity,La and Oatoms are omitted in the structu res.Blue ball,Cr atom;red ball,Mn atom.
LMCO的介电常数ε与温度的依赖关系如图7所示.在50 K处介电常数表现出显著的异常(图7(a)),这一温度与LMCO的AFM相变温度TMn~50 K一致.同时,该处介电常数的变化不受测试频率的影响,预示着本征磁电耦合相变的发生.为了进一步研究50 K附近可能的磁有序诱导的铁电相变,我们对热释电电流IP和电极化强度P进行了测试(图7(b)和图7(c)).显然,IP和P在TMn以下均可以被极化电场E翻转,进一步证实磁电耦合的出现,与介电常数ε的测试结果一致(图7(a)).因此,在TMn处的低温铁电1相(ferroelectric 1 phase,FE1)与材料的磁有序存在着紧密的关联.另外,在高温区,极化电流IP和极化强度P从180 K附近开始出现,并在125 K附近形成一个较宽的峰(图7(b)),由于这两个特征温度与AFM相变温度TCr~150 K不同,因此,高温铁电2相(ferroelectric 2 phase,FE2)不一定代表材料的本征特性,本文中不展开详细讨论.
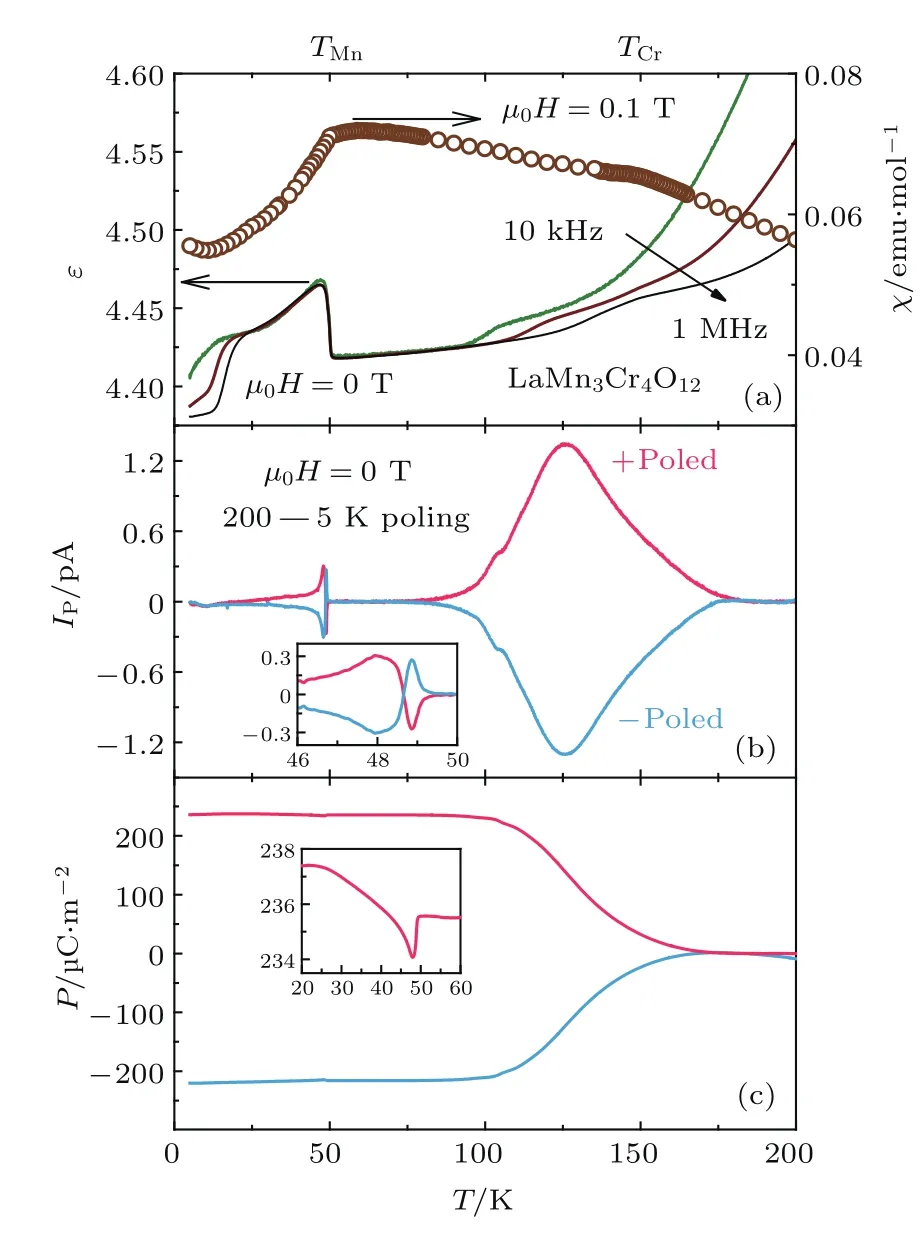
图7 (a)介电常数ε和磁化率χ,(b)热释电电流IP和(c)在正、负电极化条件下铁电极化强度P随温度的变化;ε和IP是在没有外磁场的条件下测得的,(b)和(c)中的插图给出了50 K附近的放大图Fig.7.Temperatu re dependence of(a)d ielectric constant ε and magnetic susceptibility χ,(b)py roelectric current IPand(c)ferroelectric polarization P in both+poled and−poled cond itions.ε and IPare measu red withou t magnetic fi eld.The insets of(b)and(c)showthe en larged views near 50 K.
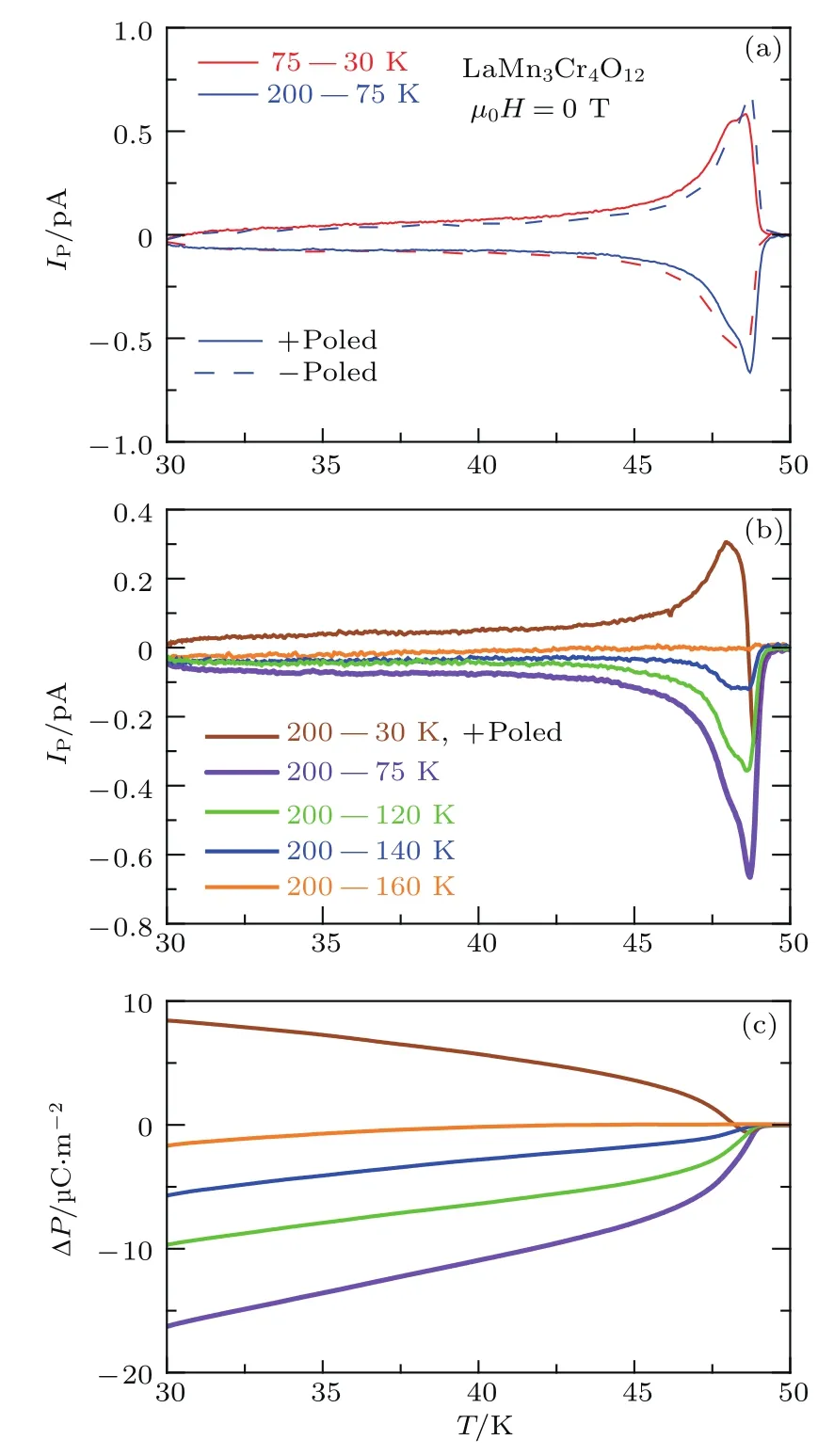
图8 TMn以下电极化行为对极化程序的依赖关系 (a)分别在200—75 K和75—30 K温度区间进行正、负电极化后,热释电电流IP随温度的变化;(b)热释电电流IP和(c)电极化的差值∆P(=P(T)−P(50 K)随温度的变化Fig.8.Poling procedu re dependence of electric behaviors belowTMn.(a)Temperature dependence of pyroelectric current IPby+poled and −poled under 200–75 Kand 75–30 Kpoling conditions.Temperatu re dependence of(b)IPand(c)the di ff erence of polarization∆P(=P(T)−P(50 K))after being+poled from200 Kdown tothe selected temperatu res.
我们注意到,在TMn处的极化电流IP并不是单个峰或谷,而是表现出反常的谷-峰特征(图7(b));同时,在TMn附近,随着温度下降,电极化P(T)先稍有减小,然后增加(图7(c)).这一行为表明此处存在着两种不同的电极化起源.为了证明这一点,我们使极化电场E分别只经过180 K和TMn处两个电极化的其中之一,得到了显著不同的结果.如图8(a)和图8(b)所示,当极化电场E只覆盖200—75 K时,极化电流IP只表现出谷的特征;当极化电场E只覆盖75—30 K时,极化电流IP只表现出峰的特征.相应的,当极化电场E只通过180 K时,Mn自旋有序导致的电极化差值(∆P=P(T)−P(50 K))为负;而当极化电场E只通过TMn时,∆P为正(图8(c)).IP(200—75 K)在TMn处的谷表明,尽管FE2相形成于高温区,但当Mn和Cr在TMn~50 K均实现自旋有序时仍然使铁电极化P发生同量级的减小.更有趣的是,IP(75—30 K)的峰表明,TMn处形成的低温FE1相独立于FE2相.由于FE1相与AFM有序密切相关,因此LMCO是一种新的自旋诱导的铁电材料.
为了进一步证明FE1相的热释电信号来源于材料本征的铁电极化而非空间电荷效应,我们用不同的外磁场对热释电电流IP和介电常数ε进行了测试.如图9(a)所示,在TMn及更低温度时,磁电及磁介电效应表现出显著的各向异性.当外磁场平行于极化电场(H//E)时,IP在TMn附近有一个量级的增加,30 K处的铁电极化的差值|∆P|也从约15µC/m2增加至约68µC/m2(图9(b));当外磁场垂直于极化电场(H⊥E)时,热释电电流IP在7 T时几乎完全被压制.

图9 50 K之下不同极化条件以及不同磁场下的(a)热释电电流IP和(b)极化差值∆P随温度的变化关系Fig.9.(a)Temperature dependence of IPand(b)the di ff erence of polarization∆P at di ff erent poling conditions and d i ff erent magnetic fi elds below50 K.
类似地,对于不同的磁场/极化场配置,介电常数ε也表现出不同的行为.当H//E时,ε在TMn处的突变随磁场的增加而更尖锐(图10(a));而当H⊥E时,ε在TMn处的突变随磁场的增加受到明显的抑制,当磁场高于7 T时几乎完全消失(图10(b)),这与上文提到的铁电极化受到磁场抑制的情形一致.另一方面,当温度高于TMn时,两种磁场/极化场配置下的IP和ε均不受磁场变化的影响,表明了FE1与FE2具有不同的铁电起源.因此,上述的测试结果表明,FE1相具有强的磁各向异性,多铁性源于LMCO特殊的自旋有序结构.
对于自旋诱导的铁电极化,目前主要有三种理论模型用于解释相关的磁电耦合多铁机理[43,44].首先是反Dzyaloshinskii-Moriya相互作用(又称为自旋流模型),其多铁性源于自旋的叉积.由于LMCO中的磁矩均为共线排列,因此这一模型无法解释LMCO的多铁性.另一种理论模型为交换收缩机理,其铁电极化来源于一对自旋的点积.并且如果这对自旋始终相互平行或反平行排列,电极化将与自旋的取向无关.而对于LMCO,根据磁点群分析和理论计算,FE1相的极化方向总是随自旋方向改变,因此交换收缩模型也无法解释LMCO磁电多铁性的成因.此外,第三种单一自旋机理(例如d-p杂化模型),也无法解释立方钙钛矿LMCO自旋诱导的多铁行为.

图10 (a),(b)分别在H//E和H⊥E 配置下,1 MHz频率和不同磁场下介电常数ε随温度的变化(为了清楚起见,对数据进行了平移)Fig.10.(a),(b)Temperature dependence of ε measured at 1 MHz and d i ff erent magnetic fi elds with H//E and H⊥E con fi gurations,respectively.The data are shifted for clarity.
从上述NPD结果可知,当 TMn<T<TCr时,Cr子晶格形成沿[111]方向的G型AFM有序(图6(a)),同时,LMCO具有Im-3的空间群,因此,该温区的磁点群为具有反演对称中心的非极化的−3′点群.当T<TMn时,Mn子晶格也形成沿[111]方向的G型AFM有序,其磁点群同样为具有反演对称中心的非极化−3点群(图6(b)).因此,单独考虑Cr,Mn子晶格,均不允许产生铁电极化.然而,当同时考虑Cr,Mn子晶格的磁点群时,整个体系将变为极化的3点群(图6(c)),从而允许沿着自旋方向的电极化的产生.因此,FE1相中的铁电极化源自Cr,Mn自旋的共同作用,LMCO成为第一个被发现的具有立方钙钛矿结构的磁电耦合多铁材料.
通过密度泛函理论(density functional theory,DFT)的计算,我们可以对FE1相中自旋诱导的铁电性有更进一步的了解.计算得到的态密度(density of states,DOS)如图11所示.可以看出,LMCO具有1.75 eV的能隙,为绝缘体.计算得到的Mn3+(3.907µB)和Cr3+(2.799µB)的自旋磁矩也与NPD的精修结果一致(表1).使用Berry相方法[45],利用实验得到的磁结构并采用弛豫方法,计算得到如下结果:1)当不考虑磁性离子的自旋轨道耦合(spin-orbital coupling,SOC)时,计算得到的FE1相的铁电极化值为0;2)当对所有自旋方向与[111]方向平行或反平行的磁性原子考虑SOC时,对于如此高对称性的结构,仍能沿[111]方向产生一个小的电极化,其大小约为3.4 C/m2.这一结果不包括离子位移的贡献,而是纯粹由电子云的空间极化导致的;3)当进一步同时考虑离子弛豫和SOC时,计算得到沿[111]方向高达7.5 C/m2的电极化值.这一计算结果与实验值(图8(c))在量级上一致,同时也与磁点群的分析得到的极化磁结构一致.
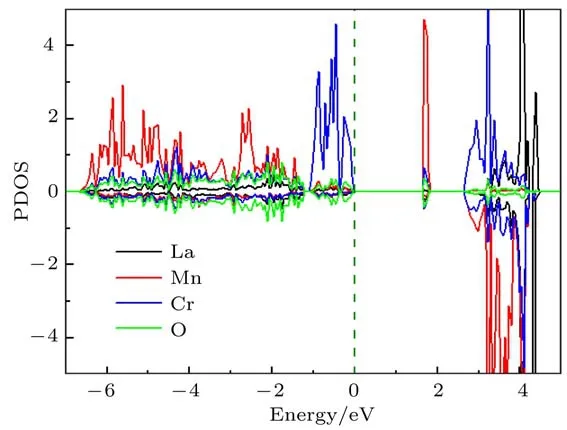
图11 不考虑SOC的双G-型AFM结构的DFT计算结果,图中给出了每种原子的投影态密度Fig.11.DFTresu lts with the dual G-type antiferromagnetism(withou t SOC).Projected density of state(PDOS)for each atomspecies is shown.
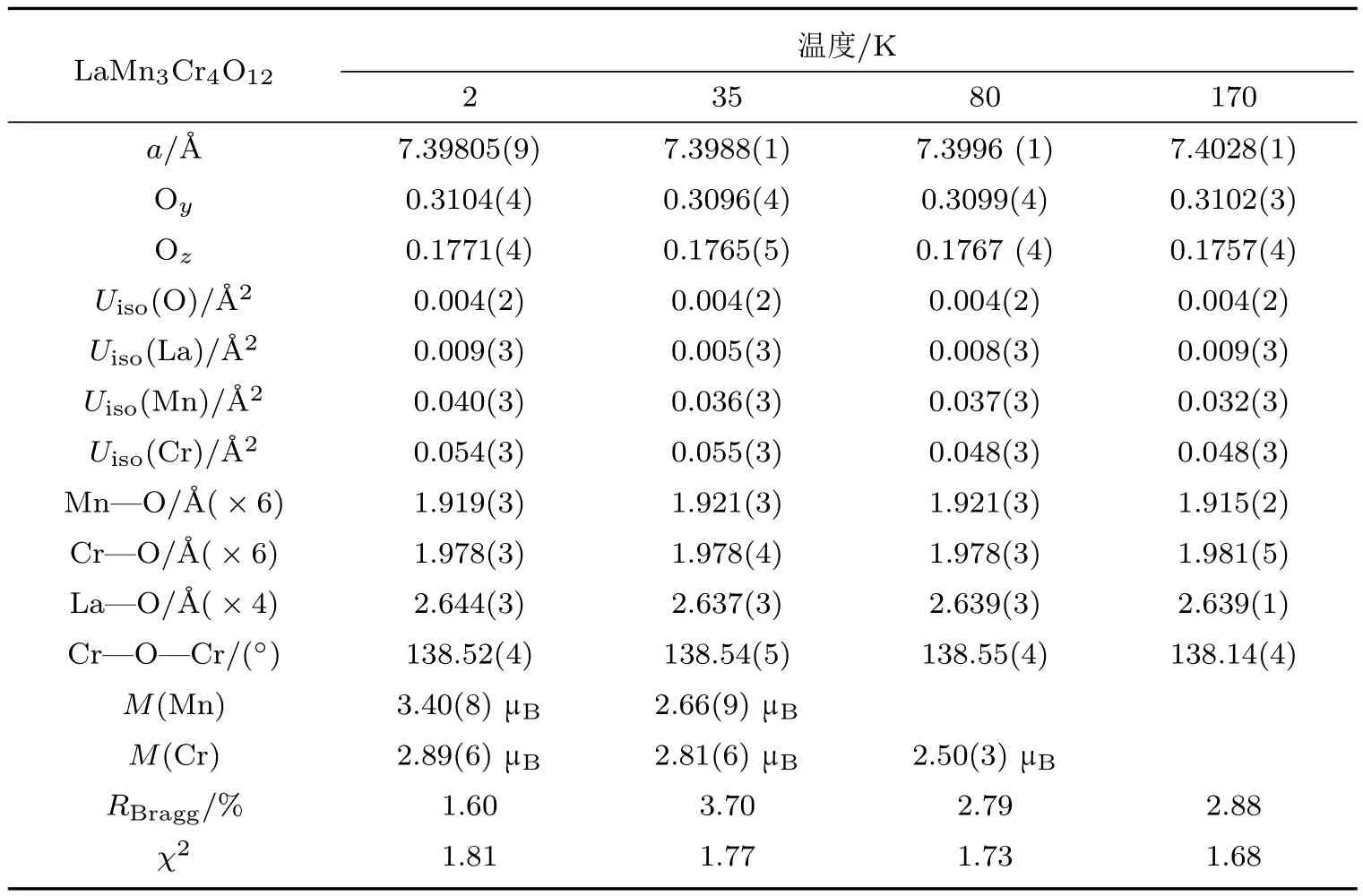
表1 LMCO不同温度下NPD数据精修给出的结构参数(空间群,Im-3(No.204);原子位置La 2a(0,0,0),Mn 6b(0,0.5,0.5),Cr 8c(0.25,0.25,0.25),O24g(0,y,z))Tab le 1.Re fi ned structu ral parameters for the NPD data of LMCOat d i ff erent temperatures.Space group,Im-3(No.204);atomic positions,La 2a(0,0,0),Mn 6b(0,0.5,0.5),Cr 8c(0.25,0.25,0.25),O24g(0,y,z).
综上所述,这项研究不仅在立方晶格多铁性材料方向取得了重要突破,而且发现了全新的多铁物理机理.DFT计算表明磁性离子的SOC效应对电极化的出现起到了至关重要的作用,但是,现有的几种磁有序产生多铁性的理论模型都不足以解释这种特殊多铁性的微观起源,需要发展全新的多铁性理论模型.此外,由于没有离子位移的贡献,该体系的电极化可能完全由电子云的偏移而产生,因此LMCO也成为研究新型电子型铁电体的典型对象.对立方钙钛矿中多铁性起源和磁电耦合机理的进一步深入探讨,将对多铁性新材料探索与新物理的研究产生深远影响.
3 单相高温亚铁磁半导体CaCu3Fe2-Os2O12
铁磁体与半导体是两种非常重要的功能材料体系,在现代工业中有着广泛的应用.如果能把这两种功能属性集成到同一单相材料中,势必为多功能自旋电子学器件的发展提供契机.但由于铁磁体和半导体在晶体结构、化学键和电子结构等方面具有本质差别,很难找到高于室温(roomtemperature,RT)的单相铁磁或亚铁磁(ferrimagnetic,FIM)半导体材料[46−49].虽然多年来研究人员对稀磁半导体的研究做出了巨大努力,但是由于其本征掺杂浓度的限制,难以找到居里温度高于RT的FM或FIM半导体.然而,通过对晶体结构和电子结构的人为设计,具有高转变温度的FM或FIM的半导体在A,B位同时有序的四重钙钛矿体系AAB2BO12(图1(d))中是有可能实现的.在这种特殊结构中,多种磁性离子(A′,B,B′)之间复杂的相互作用,导致了许多新奇的物理性质[50−52].因此,通过选取不同的磁性离子组合,一方面可调控材料的电学性质,另一方面可调控磁相互作用,从而有可能得到RT以上单相FM或FIM半导体.
我们通过高压高温的手段合成了一种新的强关联电子体系,即A,B位同时有序的四重钙钛矿结构材料CaCu3Fe2Os2O12(CCFOO)[53].该材料具有远高于RT的磁转变温度(约580 K),并且在低温下的饱和磁化强度约5µB/f.u.,预示着Cu2+(↑)Fe3+(↑)Os5+(↓)的FIM自旋排列方式. 光电流测量与DFT计算[54]表明该体系的能隙约1.0 eV,证实其具有半导体性质.因此,CCFOO作为一种少有的高温FIM半导体材料,在未来多功能自旋电子学器件开发上具有潜在应用.
图12是RT下CCFOO的XRD图谱,图中存在明显的h+k+l=奇数的特征衍射峰(例如(111),(311)等),表明B位Fe和Os的岩盐矿类型的有序排列[55].基于Rietveld方法的结构精修表明,CCFOO属于A,B位同时有序的AAB2BO12型四重钙钛矿结构,其空间群为Pn-3(图1(d)).在这个结构中,A位的Ca和A′位的Cu分别占据固定的2a(0.25,0.25,0.25)和6d(0.25,0.75,0.75)位置,而B位的Fe和B′位的Os有序地分布在4b(0,0,0)和4c(0.5,0.5,0.5)位置上.进一步的分析表明,A位的Ca和A′位的Cu几乎形成理想的1:3占位;B/B′位Fe和Os形成1:1岩盐矿的有序排列,但是Fe-Os之间存在约11%的反占位.表2列出了CCFOO的结构精修参数.根据相应的键长,用键价计算(bond valence sum,BVS)[56,57]可得到Cu,Fe,Os的价态分别为+2.11,+2.87,+5.23,给出了的价态分布.该BVS结果与后面的X射线吸收谱(X-ray absorption spectroscopy,XAS)的实验结果一致.
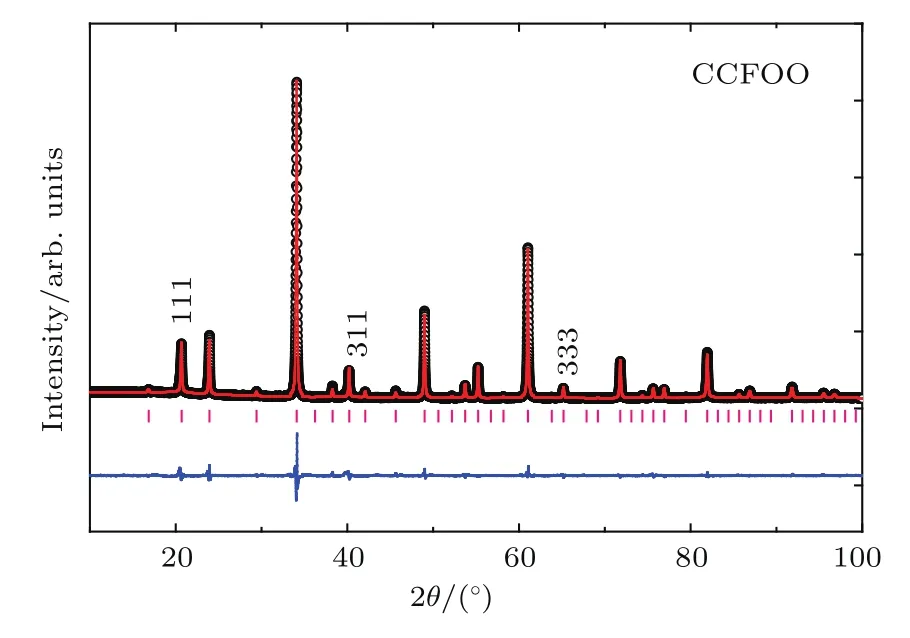
图12 RT下CCFOO的XRD图谱和结构精修结果Fig.12.XRD pattern and structu re re fi nement resu lts obtained at RTof CCFOO.
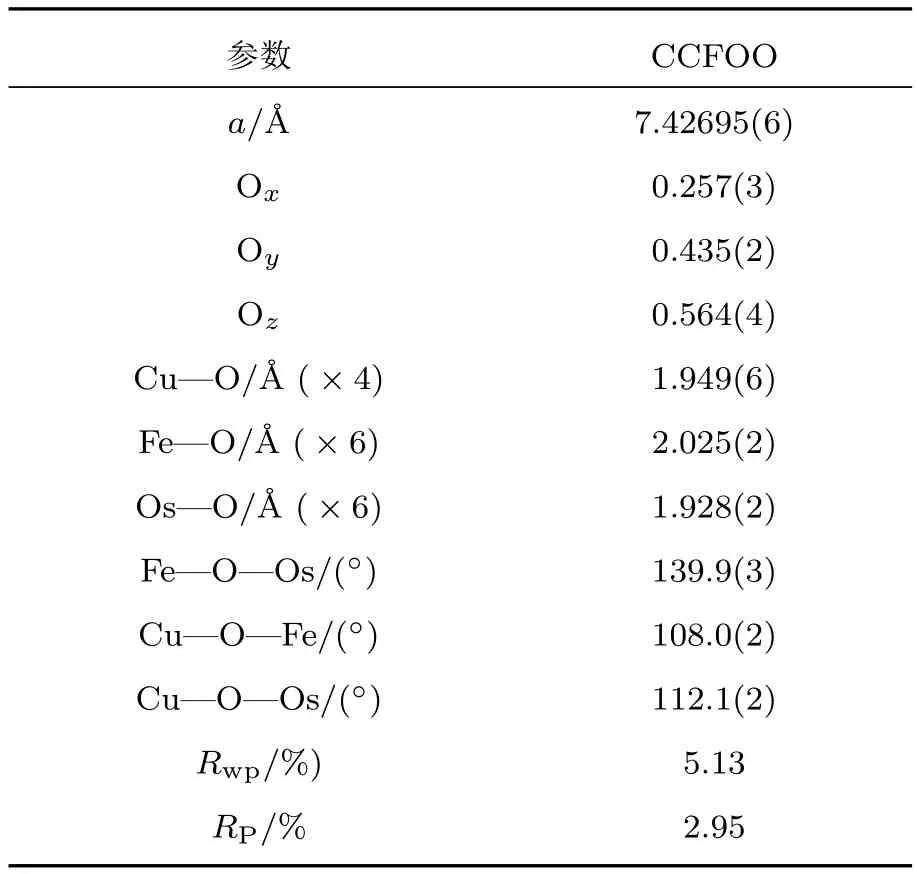
表2 RT下CCFOO的结构参数精修结果Tab le 2.Re fi ned structure parameters of CCFOOat RT.
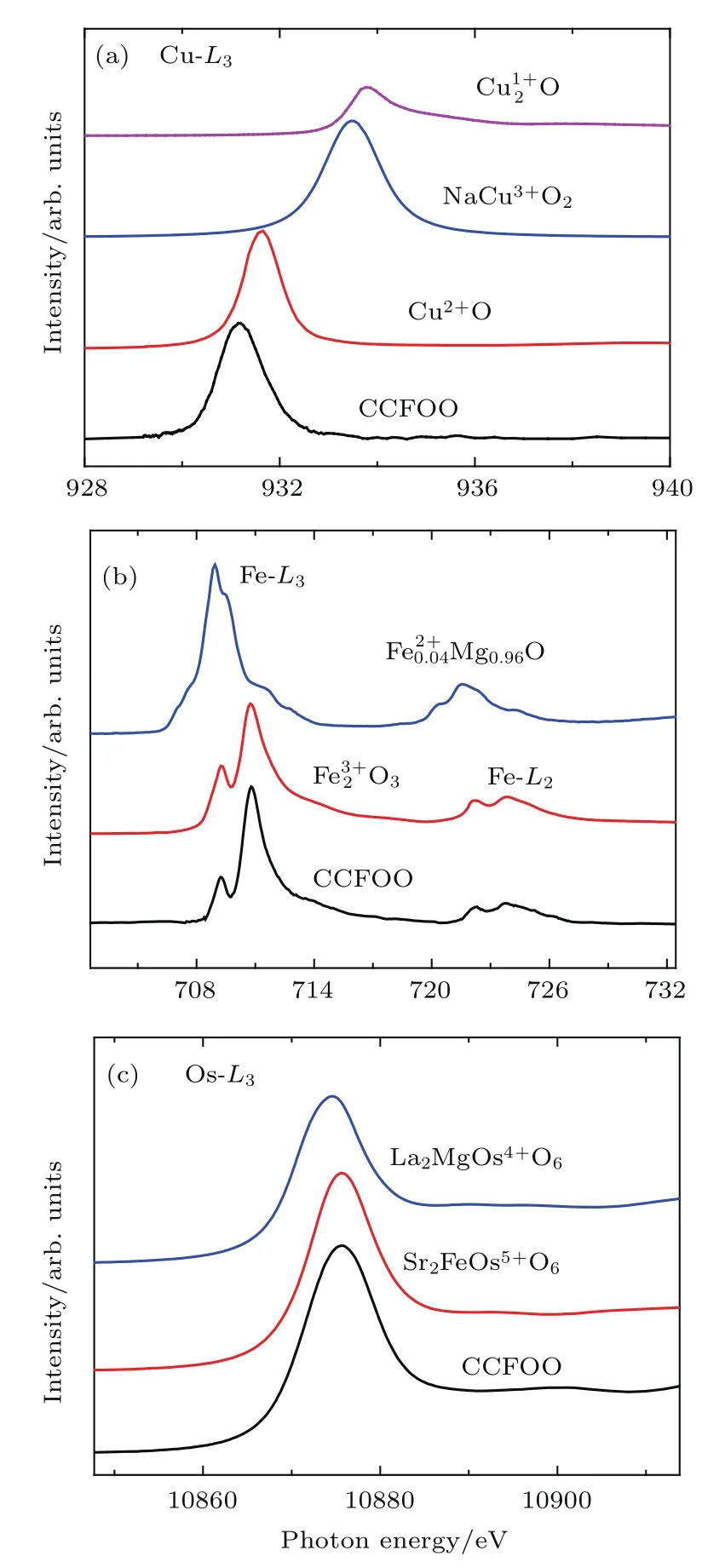
图13 (a)Cu-L2,3,(b)Fe-L2,3以及(c)Os-L3的XAS,图中给出了相关的参考样品作为对比Fig.13.XAS of(a)Cu-L2,3edges,(b)Fe-L2,3edges,and(c)Os-L3edges.The XAS of related references are alsoshown for comparison.
TM离子具有可变的化合价态,不同价态可能具有不同的磁、电特性.XAS对3d TM离子的价态十分敏感,因而是检验TM价态的有效手段.为了进一步确定CCFOO的化合价态,我们对该材料进行了XAS的测试.图13(a)给出了CCFOO的Cu-L2,3吸收边,其中Cu2O,CuO和NaCuO2作为纯的Cu1+,Cu2+,Cu3+参照物[58,59].很明显,NaCuO2的峰位比CuO高了约1.8 eV.更重要的是我们在CCFOO的Cu-L2,3吸收边能看到能量类似于Cu2+O的Cu-L2,3尖锐的的对称峰,而观察不到与Cu1+和Cu3+相关的光谱特征,因此可以确定CCFOO中Cu为+2价.图13(b)给出了Fe-L2,3吸收边,并且用Fe0.04Mg0.96O和Fe2O3作为高自旋六配位的Fe2+和Fe3+的参照物[60].可以看出,CCFOO具有和类似的光谱形状和相同能量的峰位,但是比低1.9 eV,给出了CCFOO的高自旋Fe3+的化合价.图13(c)是Os-L2,3吸收边,其中参照物为+5价的Sr2FeOsO6[61]和+4价的La2MgOsO6,容易看出CCFOO的Os-L3吸收边比La2MgOs4+O6高1.2 eV,与Sr2FeMo5+O6落在相同的能量位置,证明了CCFOO中Os5+的价态.因此,XAS清楚地表明了A,B位同时有序钙钛矿CCFOO中Cu2+,Fe3+,Os5+的价态配置.
图14为CCFOO的磁性测量结果.图14(a)为不同温度下(2,200,400 K)的等温磁化曲线,可以看出在每个测试温度都有明显的磁滞回线,表明材料具有FM或FIM性.2 K的饱和磁矩约5µB.值得注意的是,即使温度高达400 K时还有2µB的饱和磁矩,表明材料中存在强的FM或FIM磁相互作用.磁化率测试表明,随着温度降低至约580 K,CCFOO的磁化率显著增加,表明顺磁(paramagnetic,PM)向FM或FIM相变(图14(b)).因此,CCFOO具有远高于RT的居里温度(TC~580 K).
在这个复杂的A,B位同时有序钙钛矿CCFOO中,所有的TM离子Cu2+(S=1/2),Fe3+(S=5/2),Os5+(S=3/2)均可参与自旋交换相互作用.在不考虑Os5+的SOC情况下,根据局域电子模型,FM共线排列的Cu2+(↑)Fe3+(↑)Os5+(↑)和FIM共线排列的Cu2+(↑)Fe3+(↓)Os5+(↓)给出的磁矩分别为19µB/f.u.和13µB/f.u.,远大于实验观察到的5 µB/f.u.. 而Cu2+(↓)Fe3+(↑)Os5+(↑)的FIM共线排列得到的磁矩(1µB/f.u.)又过小.因此,只有Cu2+(↑)Fe3+(↑)Os5+(↓) 的FIM共线排列方式(7µB/f.u.)的饱和磁矩与实验观察到的数据最为相近.可以猜测,B/B′位Fe/Os的反占位排列以及Os-5d和O-2p轨道的强烈杂化是最可能导致实验饱和磁矩小于理想值的原因.此外,CCFOO中Cu2+(↑)Fe3+(↑)Os5+(↓)的FIM排列方式也被X射线磁圆二色(X-ray magnetic circular dichroism,XMCD)光谱所证实.如图14(c)和图14(d)所示,Fe和Cu-L2,3吸收边相同的XMCD光谱符号揭示了A′位Cu2+和B位Fe3+之间的FM排列,这与推测的Cu2+(↑)Fe3+(↑)Os5+(↓) 的FIM自旋排列相一致.

图14 (a)不同温度下磁化率随磁场的变化;(b)0.1 T下零场冷(zero- fi eld cooling,ZFC)和场冷( fi eld cooling,FC)模式下的磁化率随温度的变化;(c),(d)Fe-L2,3和Cu-L2,3的XMCD.X光的偏振方向分别平行(µ+黑线)和反平行(µ−红线)于外加磁场方向.蓝线是差值曲线Fig.14.(a)Field dependent magnetization measu red at d i ff erent temperatures;(b)temperature dependent magnetic susceptibility measured at 0.1 Twith ZFC and FC modes;(c),(d)XMCD for Fe-and Cu-L2,3edges.The photon spin is aligned parallel(µ+b lack line)and antiparallel(µ−red line)tothe applied magnetic fi eld,respectively.The di ff erence spectra are shown in b lue.
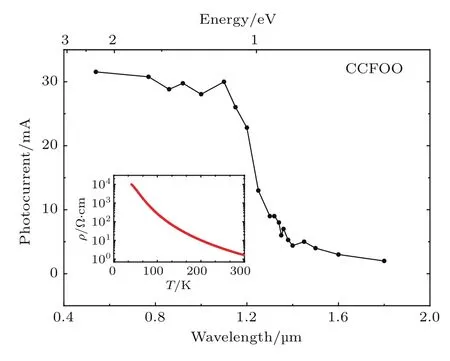
图15 RT下不同波长的光辐照时CCFOO被激发的光电流信号,插图给出了电阻率随温度的变化Fig.15.Photocu rrent signals excited by light rad iation with di ff erent wavelengths in CCFOOat RT.The inset shows the temperatu re dependence of resistivity.
我们用多晶的CCFOO样品进行了电输运测量.为了减小晶界效应,我们把多晶块体在高压下(约6 GPa)进行处理.如图15中插图所示,随着温度的降低,样品电阻逐渐增大,在低温下达到了104Ω.cm量级,表明该材料具有半导体/绝缘体性质.为了准确地确定该材料的能隙,我们进一步在RT下做了光电流的测量(图15).随着光波长的减小,在约1350 nm时,光电流信号突然增加,说明在RT下,CCFOO多晶材料的能隙约为0.92 eV.
为进一步理解CCFOO的电子结构,我们进行了相应的DFT计算,并且用广义梯度近似(general gradient approximation,GGA)和GGA+U(U表示电子关联能;Fe,Ueff=5 eV;Cu,Ueff=4 eV)的自旋极化方法来确定不同的磁基态的能量.不论是GGA还是GGA+U的方法,磁基态总是收敛成为Cu2+(↑)Fe3+(↑)Os5+(↓)的FIM有序,与实验结果相一致.DFT计算给出的总磁矩为7.01µB/f.u.,其中Cu,Fe,Os的磁矩分别为0.627,4.043和−1.421µB.当考虑SOC效应的影响时,计算结果仅发生细微改变,Cu,Fe,Os的磁矩分别变为 0.603,4.013和−1.315µB,总的磁矩值为7.19µB/f.u..图16是计算得到的电子能带结构和局部的DOS.计算揭示了CCFOO的半导体属性:基态时,自旋向上的能带有约1.8 eV的能隙,而自旋向下的能带的能隙约为1.0 eV.计算结果与光电流测量结果一致,表明了材料半导体的本征性质.另外,计算表明在B位有序钙钛矿Ca2FeOsO6与A,B位同时有序钙钛矿CaCu3Fe2Os2O12中,Fe-Os之间的磁交换相互作用强度非常近似,因此不能作为CCFOO具有高的居里温度的主导因素.然而,A′位Cu2+离子的引入可产生较强的Cu-Fe以及Cu-Os磁相互作用,因而使得CaCu3Fe2Os2O12的FIM居里温度(TC~580 K)远高于Ca2FeOsO6的FIM居里温度(TC~320 K)[53].由此可见,在A位引入额外的磁性离子从而增加体系总的磁相互作用强度,是提高有序钙钛矿磁相变温度的有效途径.该方法可用来设计室温以上磁电多功能材料.
综上所述,我们通过高压高温合成手段,首次制备了新型的A,B位同时有序的四重钙钛矿CCFOO.结构精修表明该化合物具有立方晶系,空间群为Pn-3,XAS确定了Cu2+/Fe3+/Os5+的电荷组态;磁化率测量表明CCFOO具有高达580 K的居里温度,低温下具有5µB/f.u.的饱和磁矩,DFT计算和XMCD实验结果证实了Cu2+(↑)Fe3+(↑)Os5+(↓)的FIM耦合方式;电输运测量表明该体系具有半导体的电导行为,光电流测试与DFT计算证实其能隙约为1 eV的半导体性质.因而,CCFOO提供了一个少有的既具有远高于RT的FIM居里温度,又具有较大能隙的单相半导体材料,其独特的高温FIM半导体性质在未来先进自旋电子学器件中有潜在应用.
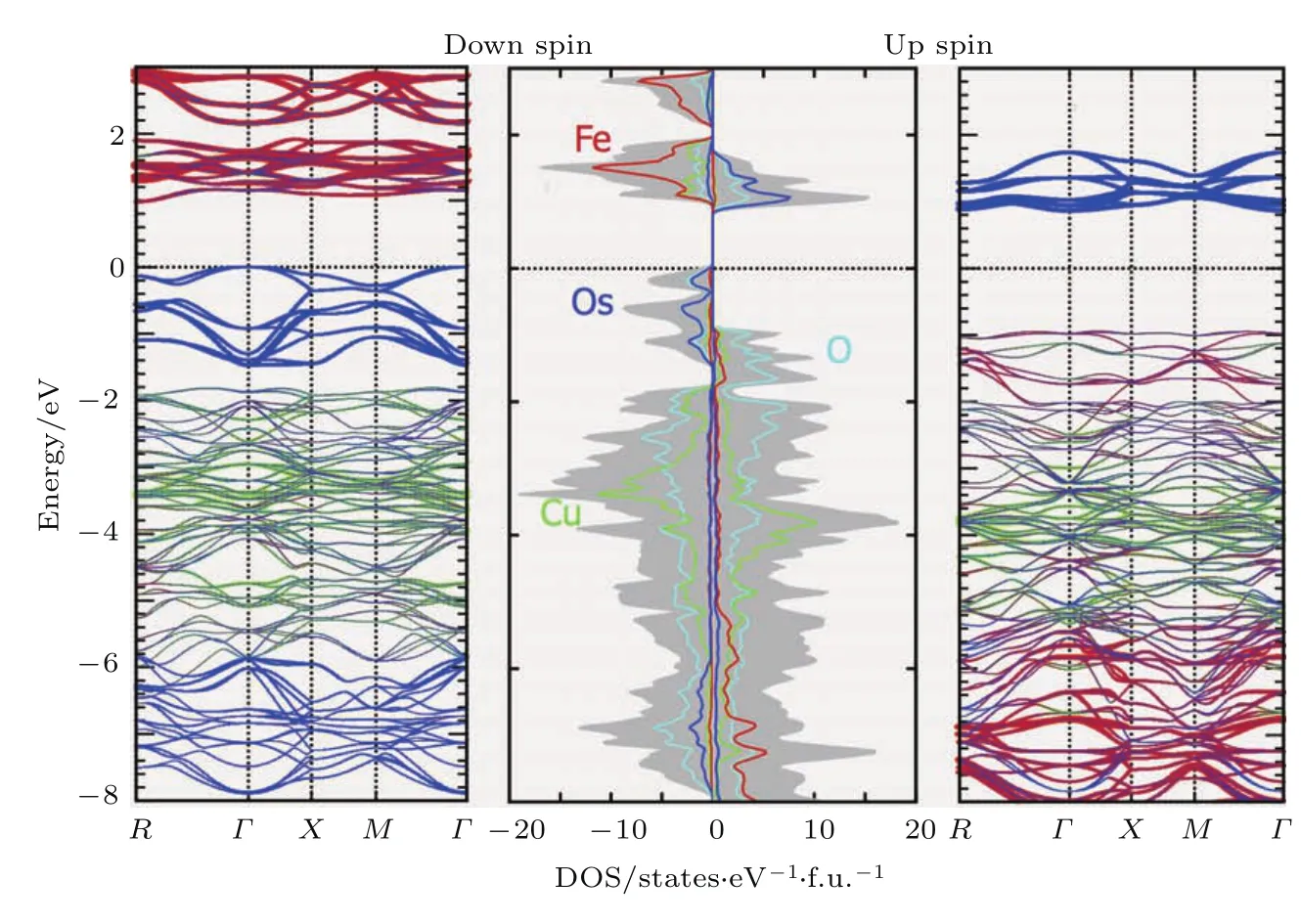
图16 CCFOO的第一性原理给出的能带结构以及局部的DOS结果Fig.16.First-principles numerical resu lts for the band structu res and partial DOS of CCFOO.
4 LaMn3Ni2Mn2O12中受A位磁性离子调控的B位正交自旋有序结构
LMNMO可以在8 GPa,1400 K的高压高温条件下合成.图17给出了LMNMO在RT下的粉末XRD、高分辨选区电子衍射(selected area electron diff raction,SAED)以及 300 K的NPD.从图17(a)SAED图样中明显的(111)超结构衍射斑点以及图17(b)NPD中强的(111)超结构衍射峰可以判定LMNMO中B位Ni和B′位Mn以岩盐矿有序的形式排布.XRD和NPD的衍射结果可以很好地用空间群为Pn-3的A,B位同时有序立方钙钛矿AAB2BO12结构来精修.其中A位的La和A′位的Mn分别占据2a(0.25,0.25,0.25)和6d(0.25,0.75,0.75)位置并形成1:3有序;B位的Ni和B′位的Mn分别占据4b(0,0,0)和4c(0.5,0.5,0.5)位置并形成1:1岩盐矿有序;O原子占据24h(x,y,z)的位置(图1(d)).表3和表4给出了精修结果.表3中2a位La,6d位Mn和4c位Mn以及24h位O的原子占据率接近100%,仅4b位上的Ni被大约10%的Mn所取代.因而,LMNMO样品满足很好的化学计量比.
为了进一步确定LMNMO的低温晶体结构,分别在200,40和3 K进行了变温NPD.NPD精修结果表明,LMNMO在温度降低至3 K时仍然保持Pn-3空间群,晶体结构未发生变化 (见表3).表4给出了各A′—O,B/B′—O键键长和键角的精修结果. 其中B/B′位的Ni—O—Mn键角约为138.5◦,表明LMNMO中NiO6和MnO6八面体存在严重的倾斜. BVS[56,57]给出了的价态组合.从BVS计算结果可以看出,LMNMO的A′位被Jahn-Teller离子占据,B位和B′位分别被非Jahn-Teller离子和占据.
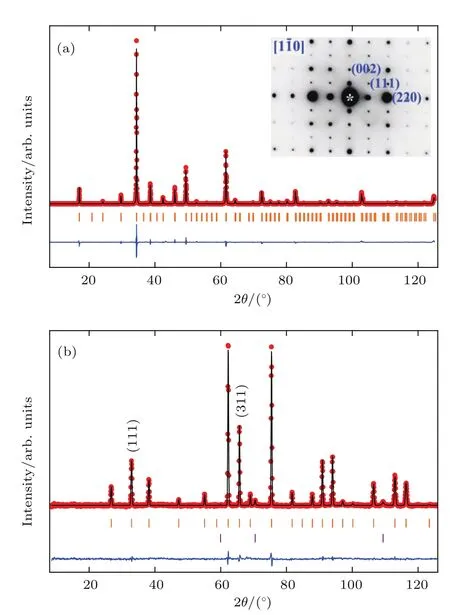
图17 LMNMO(a)在RT收集的XRD图谱和(b)在300 K收集的NPD的图谱以及相应的Rietveld精修结果,图中给出了观测值(红色圆圈)、计算值(黑线)以及差值(蓝线);上面的竖线表示允许的Bragg反射,(b)图中下面的竖线表示少量的NiO杂相(<1.2 wt%),(a)图中的插图给出了RT下沿着[10]晶带轴的选区电子衍射图案Fig.17.Rietveld refinements for(a)XRD pattern collected at RTand(b)NPD pattern at 300 Kfor LMNMO.The observed(red circles),calcu lated(b lack line),and d iff erence(b lue line)are shown.Allowed Bragg refl ections are indicated by ticks(top).The lower ticks shown in panel(b)present the small amount of impurity phase N iO(<1.2 wt%).The inset in panel(a)shows an SAED pattern along[10]zone axis taken at RT.
为了进一步验证BVS计算给出的LMNMO的价态组合,在RT下进行了XAS的研究.如图18(a)所示,LMNMO的Ni-L2,3吸收边的能量与PbNi2+O3[70]中二价Ni2+-L2,3能量重合,但是比中的三价Ni3+-L2,3的能量低1 eV,表明LMNMO中Ni以+2价存在.并且,LMNMO和PbNiO3中Ni的多重态光谱结构十分相似,表明这两个化合物存在相同的NiO6八面体配位.图18(b)给出了LMNMO中Mn-L2,3吸收边,同时给出了含有Mn3+O4平面四边形配位的A位有序钙钛矿和含有Mn4+O6八面体配位的简单钙钛矿SrMn4+O3[74]的Mn-L2,3吸收边作为对比.结果表明,Mn-L2,3的峰位逐步向高能方向由YMA移动到LMNMO,进而到SrMnO3.表明这三个化合物中Mn的价态在逐步升高.图18(b)中点线给出了YMA和 SrMnO3的Mn-L2,3吸收边以3:2的比例简单叠加.可以看出,叠加结果Mn-L2,3吸收边和LMNMO的Mn-L2,3吸收边能够很好地重合,说明LMNMO中Mn的平均价态为+3.4.从而,LMNMO的XAS证实了BVS计算给出的价态组合.
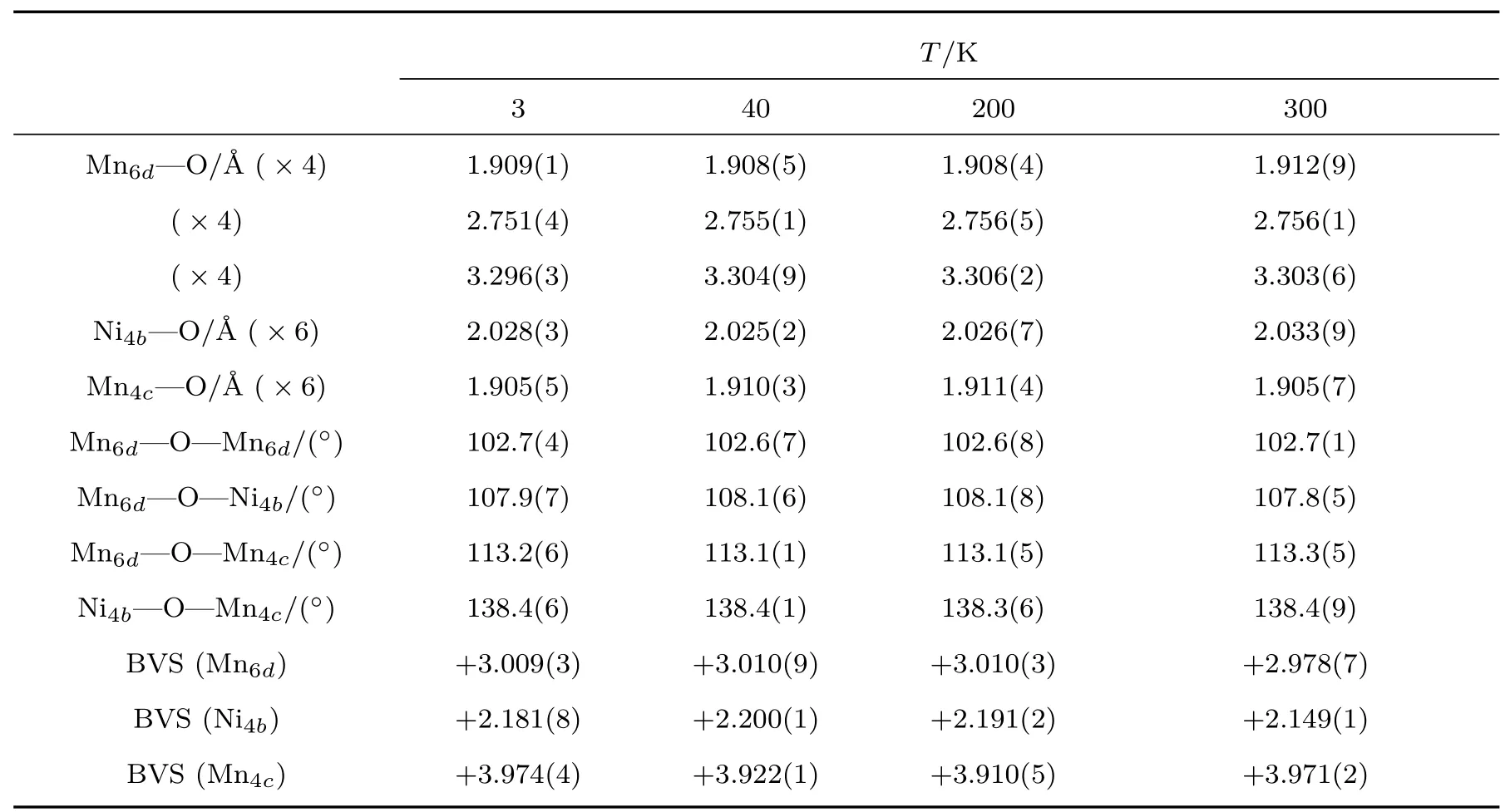
表4 从LMNMO的NPD的Rietveld精修结果中选定的键长、键角以及BVS计算结果aTab le 4.Selected bond lengths and angles and the BVS calcu lations for LMNMOobtained fromthe Rietveld refinements of NPD dataa.

图18 (a)LMNMO(红色)以及参考样品PbN i2+O3(黑色),(蓝色)的N i-L2,3吸收边的归一化XAS;(b)LMNMO(红色)以及参考样品(橙色),SrMn4+O3(蓝色)的Mn-L2,3吸收边的归一化XAS,其中,点线给出了YMn3Al4O12和SrMnO3的Mn-L2,3吸收边以3:2的比例简单叠加Fig.18. Normalized XAS of(a)N i-L2,3edges of LMNMO(red)and the reference PbN i2+O3(b lack),(b lue)and(b)Mn-L2,3edges of LMNMO(red),and the references(orange)and SrMn4+O3(b lue).The dotted line shows a simple superposition of YMn3Al4O12and SrMnO3with a 3:2 ratio.
由于LMNMO中三个不同的原子位置(即A′,B和B′位)同时被TM离子占据,因此可能产生新奇的磁性.图19(a)给出了不同磁场下LMNMO的磁化率曲线.直观上看,磁化率曲线仅仅在低温下经历了一个类似FM的转变.在0.01 T的较小外磁场下,磁化率的ZFC和FC曲线在磁转变温度之下发生了分离.然而,当用1 T的较大外磁场来克服磁畴壁能后,ZFC和FC曲线在低温下趋于重合.从图19(a)的插图可以看出,在0.01 T磁场下,磁化率曲线倒数的二阶微分存在两个明显的极值点,表明可能存在两个磁相变.为了进一步证实这两个磁相变的存在,分别测量了0 T和1 T下的比热.如图19(b)所示,零场下比热曲线分别在TN~46 K和TC~34 K处出现两个明显的 λ-型反常.当外加1 T磁场时,TN基本上保持不变,而TC明显向高温移动.基于这些特征,可以得出如下结论:LMNMO在46 K和34 K分别经历了一个长程AFM相变和一个长程FM相变.
不同温度下的磁化曲线进一步给出LMNMO中存在两个连续的磁相变的证据.如图19(c)所示,在TN之上,线性的磁化行为表明材料处于PM相.虽然在TN和TC之间没有出现典型的磁滞回线,然而40 K磁化曲线的线性外延给出了一个非零的净磁矩.由此可以推断,LMNMO在TN和TC之间存在短程FM耦合.在TC之下,出现了明显的磁滞回线,表明类FM长程序的形成.2 K的磁化曲线在高达7 T的磁场下仍然没有饱和,说明LMNMO的基态同时存在FM和AFM磁性交换作用.值得注意的是,当用线性外延扣除2 K下磁化曲线中AFM的贡献后,得到了高达约6.6µB/f.u.的饱和磁矩.在AMn3B4O12家族中,A′位Mn3+往往引入一个独立的AFM有序[42,63],而两个近邻的Ni2+(具有半满填充的轨道)和Mn4+(具有空的轨道)往往产生FM交换作用[75,76].因而可以猜测,LMNMO中类FM转变来自B/B′位Ni2+/Mn4+子晶格之间的磁交换作用.在局域电子模型下,绝缘体LMNMO中FM耦合平行排列的Ni2+和Mn4+将给出10µB/f.u.的饱和磁矩.从而,实验上观察到的约6.6µB饱和磁矩表明LMNMO可能存在一个非共线的FM自旋排列.这个假设将被后面的NPD所证实.此外,在0.01 T磁场下,ZFC和FC曲线仅在TC以下而非TN以下发生分离,同样支持B/B′位Ni2+/Mn4+非共线FM磁结构的假设.
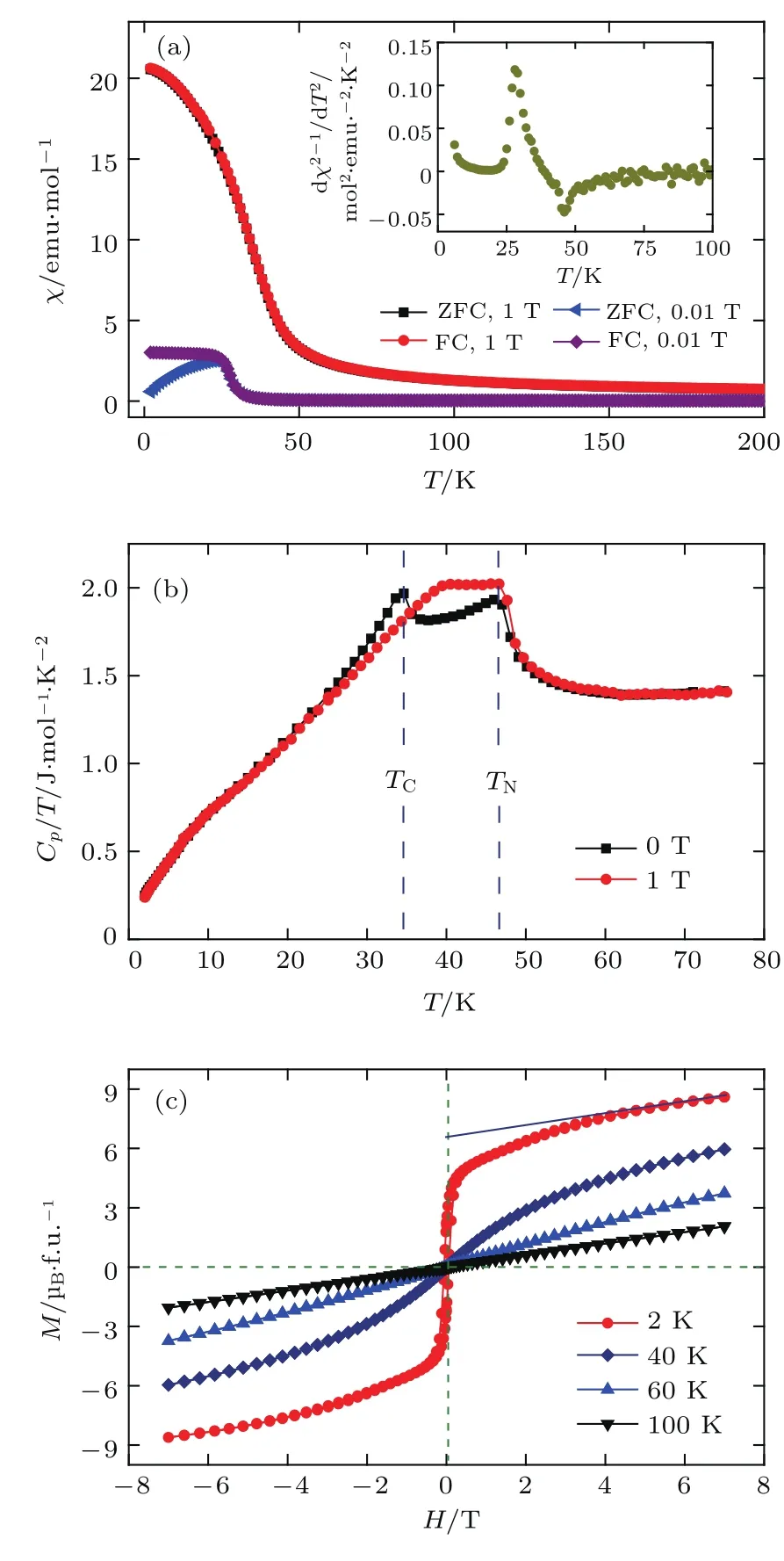
图19 (a)0.01 T和1 T磁场下磁化率随温度的变化,插图给出了0.01 T磁场下磁化率倒数的二阶微分;(b)零场和1 T磁场下比热随温度的变化;(c)不同温度下测得的磁化强度Fig.19.(a)Temperature dependence ofmagnetic susceptibility measu red at 0.01 and 1 T,the insert shows the second derivative of the inverse susceptibility at 0.01 T;(b)temperatu re dependence of specifi c heat measured at zerofield and 1 T;(c)magnetizationmeasu red at various temperatu res.
为了揭示LMNMO低温下复杂的磁性,我们对变温NPD进行了详细的磁空间群理论的分析和精修 (图20).在图20(a)中,200 K的NPD和300 K的NPD没有明显的区别.然而在TN之下40 K的衍射谱中出现了如(100)等来源于磁有序的衍射峰(图20(b)).因而,用40 K的NPD结果来研究TN和TC之间的磁结构.分析表明,该温度下表征磁结构的传播矢量k=0,因此LMNMO的晶胞和磁胞的大小一致.NPD精修结果表明,在40 K时,仅仅A′位的Mn3+离子形成了G-型AFM有序,即每个Mn3+离子和周围最近邻的Mn3+离子形成AFM排列(图22(a)).同样,LMCO[41]和YMA[77]A′位的Mn3+具有类似的G-型AFM 结构.尽管LMNMO3 K的NPD(图20(c))和40 K的NPD相比,除了磁衍射峰(如(100),(210)等)的强度有所增加外,没有其他明显的改变.然而,磁化率和比热测量结果表明,LMNMO在TC~34 K处存在一个类FM相变.对比归一化的(100)和(111)衍射峰可以看出,这两个峰的强度在TN处急剧增加,并且在TC处发生分离(图21).这些特征同样可以表明上述两个磁相变的存在.对3 K的NPD精修表明,该温度下A′位Mn3+仍然保持和40 K时同样的G-型AFM结构;此外,B位Ni2+和B′位Mn4+形成了如图22(b)所示的正交磁结构,该磁结构具有一个沿着a轴的FM分量.上述磁结构可以很好地解释LMNMO的磁化行为.因此,在TC之下,LMNMO总的磁结构由A′位Mn3+的G-型AFM结构和B/B′位Ni2+/Mn4+的正交磁结构共同组成(图22(c)).在这个自旋模型中,B/B′位Ni2+/Mn4+正交磁结构的理论净磁矩为7.07µB/f.u.,接近实验上2 K磁化曲线给出的6.6µB/f.u.,表明该模型的正确性.同样,沿着b轴和c轴分别存在两个等价磁结构模型.
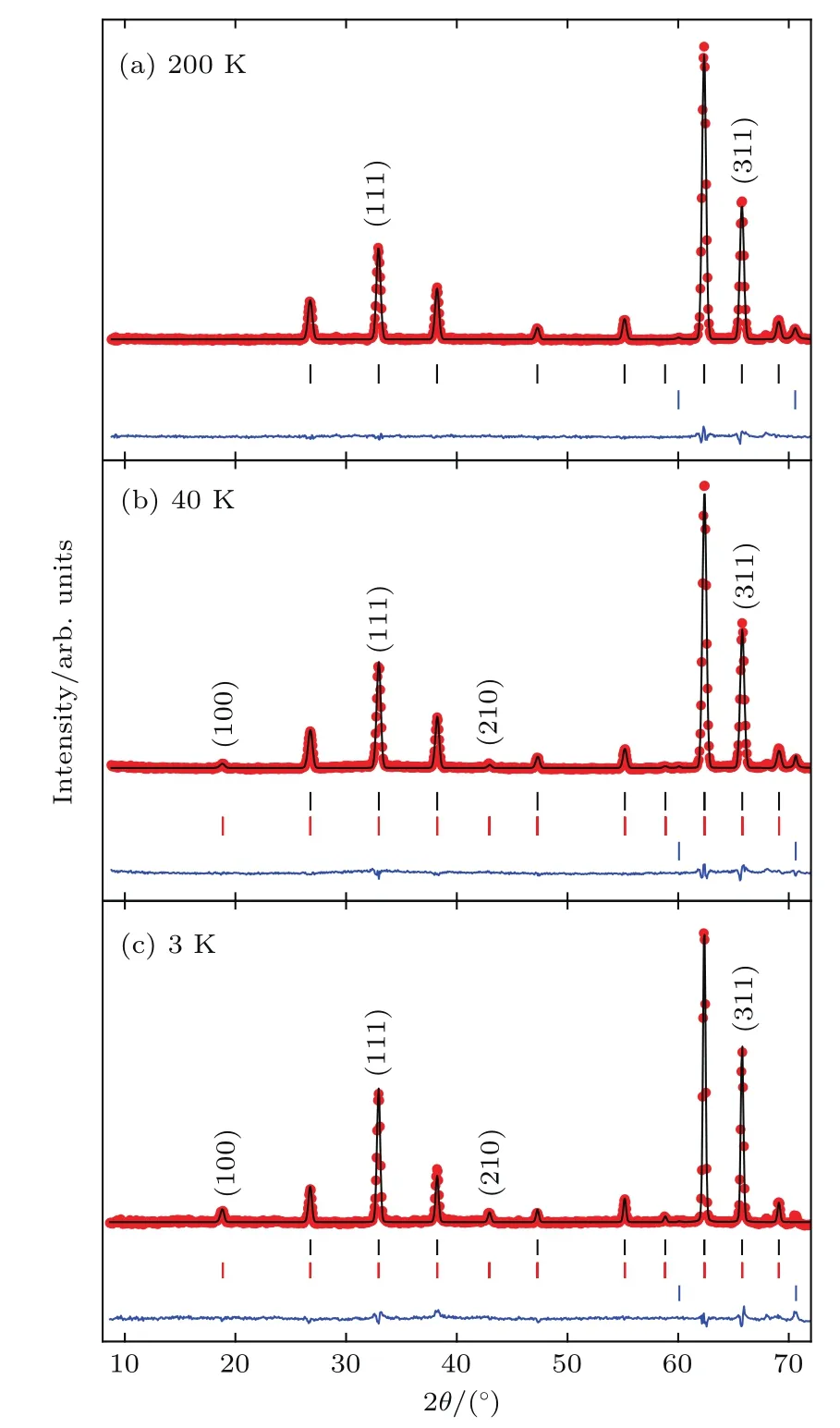
图20 LMNMO分别在(a)200,(b)40和(c)3 K下采集的NPD的Rietveld精修结果,图中给出了观测值(红色圆)、计算值(黑线)以及差值(蓝线),黑色、红色和蓝色竖线记号分别对应于允许的原子实Bragg衍射峰、磁Bragg衍射峰以及NiO杂相峰(<1.2 wt%)Fig.20.Rietveld refinements for the NPD patterns of LMNMOcollected at(a)200,(b)40,and(c)3 K.The observed(red circles),calcu lated(b lack line),and d iff erence(b lue line)are shown.The b lack,red and b lue ticks correspond tothe allowed nuclear Bragg peaks,magnetic Bragg refl ections and the impu rity phase N iO(<1.2 wt%),respectively.
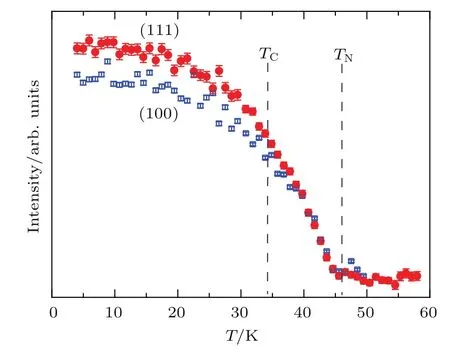
图21 归一化的(100)和(111)NPD峰的强度随温度的变化,两条曲线在TN处显著上升,并且在TC处趋向于彼此分开Fig.21.Normalized neu tron d iff raction intensity as a function of temperature for(100)and(111)peaks.These twocurves sharply increase at TNand then tend toseparate for each other belowTC.
值得注意的是,3 KNPD精修结果给出的A′位Mn3+(S=2),B位Ni2+(S=1)和B′位Mn4+(S=3/2)的自旋磁矩分别为2.94(8)µB,0.65(3)µB和0.44(3)µB.这些磁矩实验值(特别是B位Ni2+和B′位Mn4+的值)远远低于仅考虑自旋分量时离子磁矩的理论值.这个结果可以用B/B′位上轻微的原子无序以及该自旋体系中强的自旋阻挫来解释. 此外,LMNMO中引起类FM有序的Ni2+—O—Mn4+交换路径的键角不符合A2NiMnO6家族中基于Ni2+—O—Mn4+键角的磁性演变规律.如图23,LMNMO中Ni2+—O—Mn4+的键角(~ 138.5◦)落在了A2NiMnO6(A=Sc,In,Y,Bi或者稀土元素)[76,78−85]家族中AFM 对应的键角区间.这个反常表明LMNMO中A′位Mn3+可能在B/B′位Ni2+/Mn4+正交磁结构的形成中起着重要的作用.

图22 (a)A′位Mn3+(灰色)的G-型AFM结构;(b)由B位Ni2+(红色)和B′位Mn4+(蓝色)构成的正交自旋有序结构;(c)A′位,B位和B′位磁性离子构成的总的自旋结构示意图;为了清楚起见,结构中省去了O和La原子Fig.22.Schematic viewof(a)the G-type AFMstructu re of the A′-site Mn3+(gray),(b)the orthogonally ordered spin structu re composed of the B-site N i2+(red)and the B′-site Mn4+(b lue),(c)the total spin structu re composed of the A′-,B-and B′-site magnetic ions.For clarity,Oand La are omitted in the structures.
为了从根本上理解LMNMO中不同原子位置上TM离子Mn3+,Ni2+和Mn4+之间特定的自旋交换作用,利用3 KNPD给出的晶体结构参数进行了DFT计算.图24(a)给出了LMNMO中8条最重要的磁交换路径.其中,分别对应于不同路径的有效交换积分理论值(其中正值代表FM交换;负值代表AFM交换).从计算结果可以看出,对于A′位平面配位的Mn3+,最近邻(NN)、次近邻(NNN)和第三近邻(TNN)都支持AFM交换作用.相反,对于B/B′位八面体配位的Ni2+/Mn4+而言,都表现为FM交换作用.这些计算结果与磁性测量以及NPD给出的磁结构结果一致.然而,不同于LMCO(其中A′位Mn3+和B位Cr3+之间的交换作用可以忽略)的是,LMNMO表现出强的位间的交换作用.并且,交换积分在量级上与NN或NNN的和相当,从而强烈表明,LMNMO中A′位Mn3+在B/B′位Ni2+/Mn4+正交磁结构的形成起着重要的作用.此外,图24(b)中DOS计算结果给出了一个约1.5 eV的能隙,这与共顶点连接的高自旋的Ni2+O6和 Mn4+O6八面体所期望的结果一致.此外,在LMNMO中,A′位Mn3+,B位Ni2+和B′位Mn4+的DOS存在很大的重叠,这也帮助解释了A′位和B/B′位离子之间存在强的磁交换作用的原因.值得注意的是,LMNMO中Mn3+,Ni2+和Mn4+之间量级相当的交换作用可以形成强的几何磁阻挫(图24(a)).其中FM和AFM交换作用的相互竞争可能是导致LMNMO中B/B′位正交磁结构形成的主要原因[86,87].此外,LMNMO磁结构中强的磁阻挫最有可能解释NPD观测到的磁有序结构中小的自旋磁矩,即由于磁阻挫的存在抑制了磁长程有序的完全形成.这种情况在烧绿石结构中普遍存在[88].
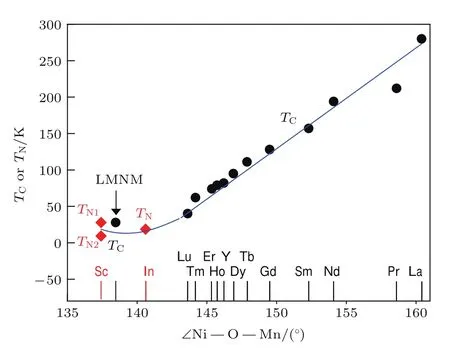
图23 A2NiMnO6家族以及LMNMO的磁有序温度随平均的N i—O—Mn键角的变化,其中,TN表示AFM奈尔温度(红色菱方);TC代表FM居里温度(黑色圆);蓝线起到对视线的引导作用Fig.23.Magnetic ordering temperature as a function of the average N i—O—Mn bond angle in A2NiMnO6family and LMNMO.TN,the AFMNéel temperature(red d iamonds);TC,the FMCurie temperatu re(b lack circles).The b lue line is a guide for the eyes.
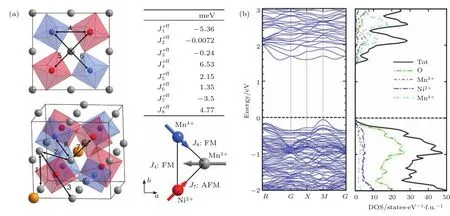
图24 (a)A′位Mn3+(灰色),B位N i2+(红色)和 B′位Mn4+(蓝色)离子之间磁交换路径以及磁阻挫模型示意图,图中给出了理论计算得到的磁交换积分常数;(b)计算得到的总的以及原子分辨的DOSFig.24.(a)Schematic viewof themagnetic exchange pathways and magnetic frustration model among the A′-site Mn3+(gray),B-site Ni2+(red)and B′-site Mn4+(b lue)ionswith the exchange constants determined fromthe theoretical calcu lations;(b)total and atom-resolved DOS fromcalcu lations.
综上所述,我们利用高压高温实验条件首次获得了A,B位同时有序钙钛矿材料该氧化物属于立方Pn-3空间群.由于LMNMO中A′位、B位和B′位之间存在复杂的磁交换作用,导致A′位Mn3+和B/B′位Ni2+/Mn4+分别在TN~46 K和 TC~34 K形成了G-型AFM和正交磁有序结构.DFT计算表明A′位和 B/B′位之间存在强的磁阻挫,从而给出了LMNMO中正交性磁结构形成的原因.从而,LMNMO成为了第一个由A′位磁性离子调控的B/B′位具有正交磁有序结构的四重钙钛矿.
5 结 论
总之,压力作为一个平行于温度与化学组分的决定物质热力学状态的重要参数,高压制备也已成为新型关联电子体系的重要源泉.由于钙钛矿灵活多变的电荷组态与晶体构型,高温高压特别适合合成ABO3钙钛矿以及A位与/或B位有序钙钛矿.在有序钙钛矿中,多个原子位置容纳过渡金属离子,因此除了传统的B位相互作用外,也存在A位相互作用以及A-B位间的相互作用,由此导致众多新颖有趣物理现象与功能属性的出现.本文中论及的A位有序钙钛矿LaMn3Cr4O12以及A,B位同时有序钙钛矿CaCu3Fe2Os2O12与LaMn3Ni2Mn2O12均是高温高压极端条件下的产物.在不同温度下,LMCO始终保持立方钙钛矿结构,空间群为Im-3.虽然这种具有反演对称中心的物质在晶体结构上不支持铁电极化,然而由A′位Mn3+与B位Cr3+磁性离子共同组成的特殊自旋结构可以打破空间反演对称,从而诱导电极化.不同于传统的铁电相变,LMCO的电极化来自于纯电子的贡献,具有较大的磁电耦合效应.LMCO是第一个被发现的具有立方晶系的钙钛矿多铁性材料,为新型多铁材料的探索以及多铁机理的研究提供了范例.半导体材料与(亚)铁磁材料是研究与应用非常广泛的两类材料体系,然而如何把半导体与铁磁这两种属性集成到一个单相材料体系中,却面临巨大的挑战.虽然通过磁性离子掺杂可获得很多稀磁半导体,但其铁磁居里温度往往较低,不利于实际应用.我们把磁性离子Cu2+引入到钙钛矿的A位,首次在高压下获得了CCFOO.Cu2+离子的引入在原有Fe-Os磁相互作用基础上增加了额外较强的Cu-Os与Cu-Fe磁相互作用,因此CCFOO具有远高于室温的亚铁磁居里温度(580 K).同时,该体系展示了半导体电输运行为,室温能隙约1 eV.可见,CCFOO是一个少有的新型单相高温亚铁磁半导体,为未来先进多功能自旋电子学器件的开发提供了重要候选材料.以往研究表明,A′位由Mn3+组成的有序钙钛矿,A′位Mn3+子晶格和B位磁性子晶格往往形成两个独立的自旋有序相,很少存在A′-B位间的磁相互作用.然而,在我们获得的多重有序钙钛矿相LMNM中,除了发现两个磁相变外,也存在较强的A′-B磁交换相互作用,这种新颖的相互作用导致了B位罕见的正交磁结构的出现,使整个体系表现出类铁磁性质.因此,LMNM成为第一个A′位由Mn3+组成的且具有铁磁行为的有序钙钛矿.
众所周知,利用常规条件已越来越难获得新材料,但高压下新材料的发现正不断涌现.而且,高压有利于稳定某些特殊晶体结构以及一些反常化合价态,这些新的体系往往具有新奇的物理性质与机理.高压作为一门材料、物理、化学等交叉的学科,尚存在巨大的未知空间等待发掘.因此,我们希望更多的年轻学者加入到这个研究领域,共同探索与开发奇妙的高压世界.
感谢中国科学院物理研究所孙阳研究员、杨义峰研究员、柴一晟副研究员以及东南大学董帅教授的合作与有益讨论.
[1]Fu HX,Cohen R E 2000Nature403 281
[2]Eitel R E,Randall C A,Sh rou t TR,Rehrig PW,Hackenberger W,Park S E 2001Jpn.J.Appl.Phys.40 5999
[3]Cox D E,Noheda B,Shirane G,Uesu Y,Fu jishiroK,Yamada Y 2001Appl.Phys.Lett.79 400
[4]Panda P K2009J.Mater.Sci.44 5049
[5]Cohen R E 1992Nature358 136
[6]Bersuker IB1966Phys.Lett.20 589
[7]GotoT,Kimu ra T,Lawes G,Ramirez AP,Toku ra Y 2004Phys.Rev.Lett.92 257201
[8]Bednorz J G,Müller KA1988Rev.Mod.Phys.60 585
[9]X iaoG,Cieplak MZ,Gav rin A,Streitz F H,Bakhshai A,Chien C L 1988Phys.Rev.Lett.60 1446
[10]Cava R J,Batlogg B,Krajewski J J,FarrowR,RuppJr L W,W hite AE,Short K,Peck W F,Kometani T1988Nature332 814
[11]MaenoY,HashimotoH,Yoshida K,Nishizaki S,Fu jita T,Bednorz J G,Lichtenberg F 1994Nature372 532
[12]Helmolt R V,W ecker J,Holzapfel B,Schu ltz L,Samwer K1993Phys.Rev.Lett.71 2331
[13]MoritomoY,Asamitsu A,Kuwahara H,Tokura Y 1996Nature380 141
[14]Toku ra Y,Tomioka Y,Kuwahara H,Asamitsu A,MoritomoY,Kasai M1996J.Appl.Phys.79 5288
[15]Toku ra Y 2006Rep.Prog.Phys.69 797
[16]Fiebig M2005J.Phys.D:Appl.Phys.38 R123
[17]Eerenstein W,Mathur N D,Scott J F 2006Nature442 759
[18]Ramesh R,Spald in N A2007Nat.Mater.6 21
[19]Spald in N A,Cheong S K,Ramesh R 2010Phys.Today63 38
[20]Mackenzie AP,Ju lian S R,D iver AJ,McMu llan G J,Ray MP,Lonzarich G G,MaenoY,N ishizaki S,Fu jita T1996Phys.Rev.Lett.76 3786
[21]Hwang HY,Iwasa Y,KawasakiM,Keimer B,Nagaosa N,Toku ra Y 2012Nat.Mater.11 103
[22]Calder S,Garlea V O,McMorrowD F,Lumsden MD,Stone MB,Lang J C,KimJW,Sch lueter J A,Shi Y G,Yamau ra K,Sun Y S,Tsu jimotoY,Ch ristianson AD 2012Phys.Rev.Lett.108 257209
[23]Carter JM,Shankar V V,Zeb MA,Kee HY 2012Phys.Rev.B85 115105
[24]Yan BH,Jansen M,Felser C 2013Nat.Phys.9 709
[25]Chen Y G,Lu Y M,Kee HY 2015Nat.Commun.6 6593
[26]Kobayashi KI,Kimu ra T,Sawada H,Terakura K,Toku ra Y 1998Nature395 677
[27]Krockenberger Y,Mogare K,Reehuis M,Tovar M,Jansen M,Vaitheeswaran G,Kanchana V,Bu ltmark F,Delin A,W ilhelmF,Rogalev A,W ink ler A,Alff L 2007Phys.Rev.B75 020404
[28]Shimakawa Y,Shiraki H,SaitoT2008J.Phys.Soc.Jpn.77 113702
[29]Ramirez AP,Subramanian MA,Gardel M,Blumberg G,LiD,Vogt T,ShapiroSM2000Solid State Commun.115 217
[30]Long Y W,Hayashi N,SaitoT,Azuma M,Mu ranaka S,Shimakawa Y 2009Nature458 60
[31]Long Y W,Kawakami T,Chen W T,SaitoT,W atanuki T,Nakaku ra Y,Liu Q Q,Jin C Q,Shimakawa Y 2012Chem.Mater.24 2235
[32]Long Y W,SaitoT,Tohyama T,Oka K,Azuma M,Shimakawa Y 2009Inorg.Chem.48 8489
[33]Long Y W,Shimakawa Y 2010NewJ.Phys.12 063029
[34]Yamada I,Etani H,Tsuchida K,Marukawa S,Hayashi N,Kawakami T,Mizumaki M,Ohgushi K,KusanoY,KimJ,Tsu ji N,Takahashi R,Nishiyama N,Inoue T,Irifune Tand TakanoM2013Inorg.Chem.52 13751
[35]W ang J,Neaton J B,Zheng H,Nagara jan V,Ogale S B,Liu B,V ieh land D,Vaithyanathan V,Sch lomD G,W aghmare UV,Spaldin N A,Rabe KM,Wuttig M,Ramesh R 2003Science299 1719
[36]Kimu ra T,GotoT,Shintani H,Ishizaka K,Arima T,Tokura Y 2003Nature426 55
[37]Katsu ra H,Nagaosa N,Balatsky V 2005Phys.Rev.Lett.95 057205
[38]SergienkoIA,DagottoE 2006Phys.Rev.B73 094434
[39]SergienkoIA,Sen C,DagottoE 2006Phys.Rev.Lett.97 227204
[40]Mostovoy M2006Phys.Rev.Lett.96 067601
[41]W ang X,Chai Y S,Zhou L,CaoHB,C ruz C D,Yang J Y,Dai J H,Y in Y Y,Yuan Z,Zhang S J,Yu R Z,Azuma M,Shimakawa Y,Zhang HM,Dong S,Sun Y,Jin C Q,Long Y W 2015Phys.Rev.Lett.115 087601
[42]Long Y W,SaitoT,Mizumaki M,Agui A,Shimakawa Y 2009J.Am.Chem.Soc.131 16244
[43]Toku ra Y,Seki S,NaotoN 2014Rep.Prog.Phys.77 076501
[44]ArimaT2007J.Phys.Soc.Jpn.76 073702
[45]Iyama A,Kimura T2013Phys.Rev.B87 180408
[46]W olf S A,AwschalomD D,Buhrman R A,Daughton J M,von Molnár S,Roukes ML,Chtchelkanova AY,Treger D M2001Science294 1488
[47]AwschalomD D,Flatte ME,Samarth N 2002Sci.Am.286 66
[48]D ietl T2010Nat.Mater.9 965
[49] Žutić I,Fabian J,Das Sarma S 2004Rev.Mod.Phys.76 323
[50]Zeng Z,G reenb latt M,Sub ramanian MA,C roft M1999Phys.Rev.Lett.82 3164
[51]AlonsoJ A,Sánchez-Benítez J,de And rés A,Martínez-Lope MJ,Casais MT,Martínez J L 2003Appl.Phys.Lett.83 2623
[52]Takata K,Yamada I,Azuma M,TakanoM,Shimakawa Y 2007Phys.Rev.B76 024429
[53]Deng HS,Liu M,Dai J H,Hu ZW,KuoC Y,Y in Y Y,Yang J Y,Wang X,ZhaoQ,Xu Y J,Fu ZM,Cai JW,GuoHZ,Jin KJ,Pi TW,SooY L,Zhou G H,Cheng J G,Chen K,Oh resser P,Yang Y F,Jin C Q,Tjeng L H,Long Y W 2016Phys.Rev.B94 024414
[54]Blaha P,Schwarz K,Madsen G KH,Kvasnicka D,Luitz J 2002W IEN 2K,An Augmen ted P lane W ave P lus Local Orbitals Programfor Calcu lating Crystal Properties(V ienna:Technische Universitat W ien)
[55]Byeon SH,Lu fasoMW,Parise J B2003Chem.Mater.15 3798
[56]Brown ID,Altermatt D 1985Acta Cryst.B41 244
[57]Brese N E,O’Keeff e M1991Acta Cryst.B47 192
[58]Hollmann N,Hu Z,Maignan A,Gunther A,Jang L Y,Tanaka A,Lin HJ,Chen C T,Thalmeier P,Tjeng L H2013Phys.Rev.B87 155122
[59]Huang MJ,Deng G,Chin Y Y,Hu Z,Cheng JG,Chou F C,Conder K,Zhou J S,Pi TW,Goodenough J B,Lin HJ,Chen C T2013Phys.Rev.B88 014520
[60]Haupricht T,Su tartoR,Haverkort MW,Ott H,Tanaka A,Hsieh HH,Lin HJ,Chen C T,Hu Z,Tjeng L H2010Phys.Rev.B82 035120
[61]Pau l AK,Jansen M,Yan B,Felser C,Reehuis M,Abdala P M2013Inorg.Chem.52 6713
[62]Senn MS,Chen W T,SaitoT,García-Martín S,Attfield J P,Shimakawa Y 2014Chem.Mater.26 4832
[63]Prodi A,G ilioli E,Cabassi R,Bolzoni F,Licci F,Huang Q Z,Lynn JW,Aff ronte M,Gauzzi A,MarezioM2009Phys.Rev.B79 085105
[64]Y in Y Y,Liu M,Dai J H,Wang X,Zhou L,CaoHB,C ruz C D,Chen C T,Xu Y J,Shen X,Yu R C,AlonsoJ A,Muñoz A,Yang Y F,Jin C Q,Hu ZW,Long Y W 2016Chem.Mater.28 8988
[65]Byeon S H,Lee S S,Parise J B,W oodward P M,Hu r N H2005Chem.Mater.17 3552
[66]Byeon S H,Lee S S,Parise J B,W oodward P M2006Chem.Mater.18 3873
[67]Chen W T,Mizumaki M,SaitoT,Shimakawa Y 2013Dalton Trans.42 10116
[68]Chen W T,Mizumaki M,Seki H,Senn MS,SaitoT,Kan D,Attfield J P,Shimakawa Y 2014Nat.Commun.5 4909
[69]Prodi A,Gilioli E,Gauzzi A,Lolzoni F,MarezioM,Bolzon F,Huang Q,SsntoroA,Lynn JW 2004Nat.Mater.3 48
[70]Inaguma Y,Tanaka K,Tsuchiya T,Mori D,Katsumata T,Ohba T,Hiraki K,Takahashi T,Saitoh H2011J.Am.Chem.Soc.133 16920
[71]Hu Z,Mazumdar C,Kaind l G,de G root F MF,W arda S A,Reinen D 1998Chem.Phys.Lett.297 321
[72]Hu Z,Golden MS,Fink J,Kaind lG,Warda SA,Reinen D,Mahadevan P,Sarma D D 2000Phys.Rev.B61 3739
[73]Tohyama T,SaitoT,MizumakiM,Agui A,Shimakawa Y 2010Inorg.Chem.49 2492
[74]KimD H,Lee E,KimHW,Kolesnik S,Dab rowski B,Kang C J,KimM,Min BI,Lee HK,KimJ Y,Kang J S 2015Phys.Rev.B91 075113
[75]Azuma M,Takata K,SaitoT,Ishiwata S,Shimakawa Y,TakanoM2005J.Am.Chem.Soc.127 8889
[76]RogadoN S,Li J,Sleight AW,Sub ramanian MA2005Adv.Mater.17 2225
[77]Toyoda M,SaitoT,Yamauchi K,Shimakawa Y,Oguchi T2015Phys.Rev.B92 014420
[78]Y iW,PrincepAJ,GuoY F,Johnson R D,Khalyavin D,Manuel P,Senyshyn A,Presniakov IA,Sobolev AV,Matsushita Y,Tanaka M,Belik AA,Booth royd AT2015Inorg.Chem.54 8012
[79]W ei Y,Liang Q F,Matsushita Y,Tanaka M,Belik AA2013Inorg.Chem.52 14108
[80]Asai K,Fu jiyoshi K,Nishimori N,Satoh Y,Kobayashi Y,Mizoguchi M1998J.Phys.Soc.Jpn.67 4218
[81]Booth R J,Fillman R,W hitaker H,Nag A,Tiwari R M,Ramanu jachary KV,Gopalakrishnan J,Lofl and S E 2009Mater.Res.Bu ll.44 1559
[82]Manna K,Bera AK,Jain M,E lizabeth S,Yusu f S M,Anil Kumar P 2015Phys.Rev.B91 224420
[83]RetuertoM,Muñoz Á,Martínez-Lope MJ,AlonsoJ A,Mompeán F J,Fernández-Díaz MT,Sánchez-Benítez J 2015Inorg.Chem.54 10890
[84]Nhalil H,Nair HS,Kumar C MN,StrydomAM,E lizabeth S 2015Phys.Rev.B92 214426
[85]Sánchez-Benítez J,Martínez-Lope MJ,AlonsoJ A,García-Muñoz J L 2011J.Phys.:Condens.Matter23 226001
[86]KajimotoR,Mochizuki H,Yoshizawa H,Shintani H,Kimu ra T,Toku ra Y 2005J.Phys.Soc.Jpn.74 2430
[87]SaitoT,Toyoda M,Ritter C,Zhang S B,Oguchi T,Attfield J P,Shimakawa Y 2014Phys.Rev.B90 214405[88]Gardner J S,G ingras MJ P,G reedan J E 2010Rev.Mod.Phys.82 53
PACS:02.10.Yn,33.15.Vb,98.52.Cf,78.47.dcDOI:10.7498/aps.66.030201
High-pressu re synthesis and special physical properties of several ordered perovskite structu res∗
Yin Yun-Yu1)Wang Xiao1)Deng Hong-Shan1)Zhou Long1)Dai Jian-Hong1)Long You-Wen1)2)†
1)(Beijing National Laboratory for Condensed Matter Physics,Institute of Physics,Chinese Academy of Sciences,
Beijing 100190,China)2)(Collaborative Innovation Center ofQuantumMatter,Beijing 100190,China)(Received 17 January 2017;revised manuscript received 18 January 2017)
Strongly correlated electronic systemswith ABO3perovskite and/or perovskite-like structures have
much attention.High pressure is an eff ectivemethod toprepare perovskites,in particular A-site and/or B-site ordered perovskites.In these ordered perovskites,both Aand Bsites can accommodate transition-metal ions,giving rising tomultiple magnetic and electrical interactions between A-A,B-B,and A-Bsites.The presence of these newinteractions can induce a wide variety of interesting physical properties.In this reviewpaper,we will introduce an A-site ordered perovskite with chemical formula AAB4O12and twoA-and B-site ordered perovskites with chemical formula AAB2BO12.All of these compounds can be synthesized on ly under high pressure.In the A-site ordered LaMn3Cr4O12with cubic perovskite structure,magnetoelectricmultiferroicity with newmultiferroicmechanismis found tooccur.This is the fi rst observation ofmultiferroicity appearing in cubic perovskite,thereby opening theway toexploring newmu ltiferroicmaterials and mechanisms.In the A-and B-site ordered perovskite CaCu3Fe2Os2O12,a high ferrimagnetic Curie temperature is observed tobe around 580 K.Moreover,this compound exhibits semiconducting conductivity with an energy band gapof about 1 eV.The CaCu3Fe2Os2O12thus provides a rare single-phase ferrimagnetic semiconductor with high spin ordering temperature well above roomtemperature as well as considerable energy band gap.Moreover,theoretical calculations point out that the introducing of A′-site Cu2+magnetic ions can generate strong Cu-Fe and Cu-Os spin interactions.As a result,this A-and B-site ordered perovskite has a much higher Curie temperature than that of the B-site only ordered perovskite Ca2FeOsO6(∼320 K).In addition,we alsofor the fi rst time prepare another A-and B-site ordered perovskite LaMn3Ni2Mn2O12.In the reported ordered perovskiteswith Mn3+at the A′site,the A′-Bintersite spin interaction is usually negligib le.In our LaMn3Ni2Mn2O12,however,there exists the considerable A′-Binteraction,which is responsib le for the rare formation of B-site orthogonal spin structure with net ferromagnetic moment.
high-pressure synthesis,ordered perovskite,multiferroicity,spin ordering
10.7498/aps.66.030201
∗国家重点基础研究发展计划(批准号:2014CB921500)、国家自然科学基金(批准号:11574378)和中国科学院先导B项目(批准号:XDB07030300)资助的课题.
†通信作者.E-mail:ywlong@iphy.ac.cn
*Project supported by the National Basic Research Programof China(G rant No.2014CB921500),the National Natu ral Science Foundation of China(Grant No.11574378),and the Strategic Priority Research Programof the Chinese Academy of Sciences(G rant No.XDB07030300).
†Corresponding author.E-mail:ywlong@iphy.ac.cn
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01
航天电子对抗(2022年2期)2022-05-24
北京航空航天大学学报(2021年9期)2021-11-02
陶瓷学报(2021年2期)2021-07-21
陶瓷学报(2019年6期)2019-10-27
发明与创新·小学生(2019年11期)2019-08-11
航天电子对抗(2019年4期)2019-06-02
军事文摘·科学少年(2017年4期)2017-06-20
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
太阳能(2015年4期)2015-02-28
