
途径优化强化枯草芽胞杆菌合成肝素前体
2017-07-01 18:50:19张琳培王浩周正雄堵国成陈坚康振
生物工程学报 2017年6期
张琳培,王浩,周正雄,堵国成,陈坚,康振
途径优化强化枯草芽胞杆菌合成肝素前体
张琳培1,2,王浩1,2,周正雄1,2,堵国成1,2,陈坚1,2,康振1,2
1 江南大学工业生物技术教育部重点实验室,江苏无锡 214122 2 江南大学生物工程学院,江苏无锡 214122

张琳培, 王浩, 周正雄, 等. 途径优化强化枯草芽胞杆菌合成肝素前体. 生物工程学报, 2017, 33(6): 936–945.Zhang LP, Wang H, Zhou ZX, et al. Optimization of heparosan synthetic pathway in Bacillus subtilis 168. Chin J Biotech, 2017, 33(6): 936–945.
肝素前体是化学酶法合成肝素的起点,肝素前体的微生物高效合成具有重要意义。在已构建的产肝素前体的枯草芽胞杆菌((1.71±0.08) g/L) 中,分析了UDP-葡萄糖醛酸 (UDP-GlcUA) 途径中关键酶基因 (、、) 以及UDP-乙酰氨基葡糖 (UDP-GlcNAc) 途径中关键酶基因 (、、) 的过量表达对肝素前体产量及其分子量的影响。在此基础上,通过共表达、、、和基因,摇瓶中肝素前体产量提高至(2.89±0.11) g/L,分子量为(75.90±1.18) kDa。通过在3 L发酵罐中进行补料分批发酵,肝素前体的产量最终积累到(7.25±0.36) g/L,分子量为(46.66±2.71) kDa,为工业化生产肝素奠定了基础。
肝素前体,枯草芽胞杆菌,途径优化,产量,分子量
肝素 (Heparin,HP) 作为一种高度硫酸化的糖胺聚糖[1],在胚胎发育、免疫应答、病毒或细菌感染及细胞分化等方面发挥重要作用[2],临床上也可作为抗凝与抗血栓药物[3]。肝素主要通过动物提取法获得[2,4],然而该方法存在种间疾病感染[5]和过硫酸软骨素污染[6-7]等风险,且提取过程往往造成环境污染[3]。另外,通过化学法也可从头合成低分子量的肝素[8],但因原料昂贵、步骤繁多和产量极低等缺点难以进行大规模制备[9-10]。近年来,以肝素前体为合成起点进行硫酸化的化学酶法逐渐受到研究者的重视[1,11],因此,在微生物中实现肝素前体的高产成为了重要环节。
肝素前体 (Heparosan) 由葡萄糖醛酸 (GlcUA) 和N-乙酰氨基葡糖 (GlcNAc) 通过β-1,4和α-1,4糖苷键交替连接形成[12-13],在大肠杆菌K5和多杀巴斯德菌中天然存在[14-15]。此外,Zhang等在.BL21 (DE3) 中实现了肝素前体的高效异源合成[16],而我们团队通过共表达来自K5的与合酶基因,首次在食品级微生物枯草芽胞杆菌中获得了肝素前体[5],有效避免了致病性菌株生产肝素前体的风险。
目前,一些团队致力于代谢工程改造提高肝素前体产量。基于哺乳动物糖胺聚糖的生物合成,认为UDP-葡萄糖脱氢酶催化的UDP-Glc到UDP-GlcUA的反应为限速步骤[17],Zhang等发现过表达该酶使产量和分子量均提高[16],而Roman团队却在.K5中得到产量降低、分子量不变的不同结论[18]。Lidholt等还发现只增加UDP-GlcNAc使肝素前体合成速率降低、分子量减小,但当UDP-GlcUA和UDP-GlcNAc处于平衡时,合成速率与分子量再次增大[19]。虽然肝素前体的生物合成与分子量调控机制尚不清楚,但两前体物同合酶的结合具有竞争关系,因此它们的浓度与比例将成为关键因素[17]。
为探究合成途径中除UDP-葡萄糖脱氢酶以外的其他关键酶及代谢流量对产物合成与分子量的影响,并获得肝素前体的高产菌株,本研究在之前工作的基础上[5]进行代谢改造,强化UDP-GlcUA途经和UDP-GlcNAc途径中关键酶基因的表达,并进一步通过3 L发酵罐放大培养,使肝素前体积累量最终提高至7.25 g/L,分子量为46.66 kDa。
1 材料与方法
1.1 材料
1.1.1 菌种和质粒
所用菌株和质粒均为本实验室保存,详见表1。
1.1.2 酶、引物、DNA marker及相关试剂盒
DNA聚合酶购自宝赛生物 (杭州) 有限公司;DNA marker购自TaKaRa (大连);PCR引物 (表2) 由生工生物工程 (上海) 有限公司合成;质粒小量抽提试剂盒购自生工生物工程 (上海) 有限公司。
1.2 培养基
1.2.1 筛选培养基与种子培养基
向LB培养基 (蛋白胨 10 g/L,氯化钠 10 g/L,酵母粉 5 g/L) 中添加无水硫酸镁溶液和木糖溶液,使其在培养基中的终浓度分别为1.5 g/L和20 g/L,自然pH。制备固体平板时添加琼脂20 g/L。
1.2.2 改良发酵培养基
蔗糖 50 g/L (摇瓶培养) 或15 g/L (3 L罐培养),酵母粉 20 g/L,蛋白胨 2 g/L,硫酸镁 1.5 g/L,50 mmol/L磷酸钾缓冲液,pH为7.0[5]。

表1 本文所用的质粒与菌株

表2 本文所用的引物
1.3 培养方法
1.3.1 种子培养
挑取生长良好的单菌落接种于含25 mL种子培养基的250 mL三角瓶中,根据需要添加50mg/mL卡那霉素,37 ℃、200 r/min培养16 h。
1.3.2 摇瓶培养
将种子培养液按10%接种量转接至50 mL (摇瓶容量为500 mL) 改良发酵培养基中,根据需要添加50mg/mL卡那霉素,37 ℃、200 r/min培养48 h。在接种后第2 h添加木糖溶液使其终浓度为20 g/L进行诱导。
1.3.3 3 L罐补料分批培养
将重组菌单菌落接种至150 mL (摇瓶容量为500 mL) 种子培养基中,37 ℃、200 r/min培养16 h,将种子培养液按10%的接种量转接至含1.35 L改良发酵培养基的3 L发酵罐中,并添加50mg/mL卡那霉素。在接种后第2 h添加木糖溶液使终浓度为20 g/L进行诱导。使用5 mol/L NaOH溶液控制pH为7.0,温度为37 ℃,搅拌转速在接种后8 h内为600 r/min,8 h后为800 r/min,通气量为2.0 vvm。补料料液为800 g/L的蔗糖母液,当发酵液中蔗糖浓度低于5 g/L时开始补料,维持残糖浓度在0−5 g/L。在8−12 h期间流加速度分别为7.5、7.5、15、10 g/(L·h),此后保持5 g/(L·h) 的流速至发酵结束。
1.4 方法
1.4.1 枯草芽胞杆菌的转化及重组子的鉴定
枯草芽胞杆菌的转化采用电转方式,转化后涂布于含50mg/mL卡那霉素的筛选平板,挑选转化子,提取质粒进行PCR验证。
引物见表2。质粒pP43-D、pP43-DB、pP43-DBA、pP43-U、pP43-UM、pP43-UMS、pP43-DU和pP43-DU-PBMS分别用引物P43-F/ tuaD-R、tuaD-F/gtaB-R、gtaB-F/pgcA-R、P43-F/ glmU-R、glmU-F/glmM-R、glmM-F/glmS-R、tuaD-F/glmU-R和glmU-F/gtaB-R进行验证。
1.4.2 菌体生长密度测定
取1 mL发酵液在12 000 r/min下离心2 min。用蒸馏水重悬菌体,以蒸馏水调零,利用紫外分光光度计测定波长为600 nm处的吸光值,根据吸光度有效值范围 (0.25−0.80) 适当稀释菌液。
1.4.3 肝素前体的纯化与定量检测
取样的发酵液在10 000 r/min下离心5 min分离上清液和菌体。向上清液中加入3倍体积的无水乙醇,混匀后于4 ℃放置1 h。此后在 5 000 r/min下离心10 min,收集沉淀。向沉淀中加入等体积的蒸馏水,于4 ℃放置使沉淀重新溶解,该纯化步骤重复3次,获得肝素前体的水溶液。采用硫酸咔唑比色法定量测定肝素前体,具体参见文献[20]。利用紫外分光光度计检测波长为530 nm处的吸光值,根据吸光度有效值范围 (0.25−0.80) 适当稀释样品溶液。
1.4.4 肝素前体分子量的测定
肝素前体的质量平均分子量 (w)、数量平均分子量 (n) 和多分散性系数p(p=w/n) 采用高效排阻色谱联合多角度激光散射法 (HPSEC-MALLS) 测定。分析条件:流动相为0.1 mol/L的硝酸钠溶液,采用凝胶色谱柱Ultrahydrogel Linear (300 mm×7.8 mm;Waters Co., Milford, MA, USA),流速0.5 mL/min,柱温50 ℃,进样量20 μL,检测器为多角度激光散射检测器 (Wyatt Technology DAWN HELEOS,Santa Barbara,CA,USA) 和示差折光检测器 (Optilab Rex,Wyatt Technology Co.,USA)。
1.4.5 蔗糖的定量检测
取1 mL发酵液在4 ℃、12 000 r/min下离心2 min,保留上清液,经0.22mm过滤器过滤后,采用HPLC定量检测蔗糖[5]。分析条件:流动相为乙腈水溶液 (70∶30,/),色谱柱为NH2柱 (4 mm×250 mm),流速1.0 mL/min,柱温32 ℃,进样量10 μL,检测器为示差折光检测器 (Shimadzu RID-10 A)。
2 结果与讨论
2.1 产肝素前体途径优化重组菌的构建
图1A中,从葡萄糖到两种前体物UDP-GlcUA和UDP-GlcNAc的代谢途经基因均为.168内源性基因[21],但.168缺少以两种前体物合成肝素前体所需的糖基转移酶基因和[22-23],我们之前已通过基因组整合共表达和,实现了枯草芽胞杆菌生产肝素前体[5]。
但是,UDP-GlcUA途径中的6-磷酸葡萄糖 (Glc-6-P) 和UDP-GlcNAc途径中的6-磷酸果糖 (Fru-6-P) 还分别参与磷酸戊糖途径和糖酵解途径[21],消耗部分原料。同时两前体物UDP-GlcUA和UDP-GlcNAc既要合成目标产物,也要参与合成细胞壁,供给菌体生长的需要[17],因而制约了肝素前体的积累。为进一步提高肝素前体产量,过量表达合成途径的关键酶基因,强化UDP-GlcUA途径和UDP-GlcNAc途径代谢流,使前体物充足。

图1 肝素前体合成途径优化
将曾构建的8个过表达途径基因的重组质粒 (图1B,表1) 转化产肝素前体的.E168H[5],经PCR验证,获得了相应的8株重组菌 (表1),结果如图2所示。
图2 PCR验证重组枯草芽胞杆菌
2.2 优化肝素前体合成途径基因对产量的影响
将8株重组菌与对照菌.E168H进行摇瓶发酵,第48 h产量结果如图3所示。
从图3A看出,与对照株相比,单独上调基因的表达显著增加了肝素前体的产量,达到 (2.65±0.13) g/L,证明了UDP-葡萄糖脱氢酶 (编码) 催化的UDP-葡萄糖 (UDP-Glc) 到UDP-GlcUA的反应为肝素前体合成的一个重要限速步骤[16-17]。进一步过表达和基因上调UDP-Glc和UDP-GlcUA的供应,产量略有降低,为 (2.32±0.05) g/L,这一结果可能是由于和基因的共同强化引起Glc-6-P储量减少,继而导致UDP-GlcNAc途径代谢流向UDP-GlcUA途径转化,使KfiA转运酶获得UDP-GlcNAc成为限速因素,降低了肝素前体合成的起始速率[18]。同时,两前体物存在竞争关系,UDP-GlcUA过量时作为竞争抑制剂占据UDP-GlcNAc与KfiA酶的结合位点,使产量降低[17]。而进一步强化UDP-GlcUA的上游合成途径,同时过表达、和基因,产量((2.37±0.02) g/L) 较过表达和时没有明显变化,即对肝素前体产量无正向作用,该现象与另一种糖胺聚糖——透明质酸的合成调控一致[24],表明Glc-6-P为磷酸戊糖途径和糖酵解途径的关键节点受到严格调控。

图3 产肝素前体枯草芽胞杆菌的摇瓶培养
对于合成另一前体物的UDP-GlcNAc途径,单独上调基因的表达,肝素前体产量达到(2.06±0.10) g/L,说明UDP-N-乙酰氨基葡糖焦磷酸化酶 (编码的双功能酶) 催化的1-磷酸氨基葡糖 (GlcN-1-P) 到UDP-GlcNAc的反应为肝素前体合成的另一限速步骤。然而,过表达和时,产量无明显变化((1.98±0.04) g/L),说明6-磷酸氨基葡糖 (GlcN-6-P) 的合成受严谨调控。在此基础上进一步强化基因后,产量恢复到 (2.30±0.22) g/L,这一结果证明酰胺转移酶 (编码) 是肝素前体合成的又一重要途径酶。
另外,为了研究同时提高两条代谢通路流量对肝素前体产量的影响,过量表达了两个限速酶基因和,产量为 (2.52±0.06) g/L。而同时上调所有基因、、、和的表达,促使肝素前体的积累量达到最大,为 (2.89±0.11) g/L,提高了69.01%,说明两前体物UDP-GlcUA和UDP-GlcNAc的比例平衡对加快肝素前体的聚合速度非常有利[17,19]。并且,从图3B中看出,与对照株相比,重组质粒pP43-DU-PBMS的转入并没有成为菌体生长的负担,菌体量反而稍有提高,不同于细胞生长和肝素前体合成具有竞争关系的说法[16-17],这可能是由于增大途径代谢流获得了充足的UDP-GlcUA和UDP-GlcNAc,在满足细胞壁形成需要后更多地用于目标产物的合成。
2.3 优化肝素前体合成途径基因对分子量的影响
已有报道一些因素如代谢流分布、前体物浓度及比例等对肝素前体的分子量有影响[17-19],因此对上述8株重组菌和对照菌所产肝素前体的分子量进行了MALLS-SEC分析,结果如表3所示。
过表达和共表达-均使w增大,分别为 (67.70±1.29) kDa和 (71.40±1.82) kDa,说明UDP-GlcUA途径代谢流的增加对延长肝素前体多糖链有利,这不同于在野生菌株.K5中提高UDP-葡萄糖脱氢酶的活性,而肝 素前体分子量未受影响[18],可能是.与.对UDP-GlcUA的合成具有不同的调控机制。进一步过表达基因导致了w的降低((53.65±2.06) kDa),反映出该基因对糖链延伸有负面影响。
过表达和组合过表达、均能明显促进w的提高((61.12±0.98)−(73.83±1.65) kDa),表明UDP- GlcNAc的浓度与肝素前体分子量存在正相关性,这一现象虽与Lidholt得出的实验结论相反[19],但与透明质酸的分子量调控相似[24-26]。
此外,共表达和基因,使w提高至 (58.79±0.80) kDa,而组合共表达时,w达到最大,为 (75.90±1.18) kDa,证明前体物UDP-GlcUA和UDP-GlcNAc的量和比例是除肝素前体合酶固有性质和外部环境以外的影响肝素前体分子量的一大重要因素[17,19]。另外还发现,8株重组菌合成的肝素前体,其多分散性系数 (p,表3) 在1.09−1.42之间,尤其是当前体物平衡时,产物分子量分布范围更集中 (1.09−1.16),说明可通过途径优化实现更统一分子量的肝素前体的生产。
2.4 发酵罐分批补料培养的研究
根据摇瓶发酵结果,将重组菌.E168H/pP43-DU-PBMS在3 L发酵罐中进行放大培养。为保证碳源的充足,实现菌体高密度发酵,当残糖含量低于5 g/L时,流加800 g/L的蔗糖母液,维持残糖浓度在0−5 g/L之间。结果如图4所示,可以看出大量的肝素前体的积累主要集中于菌体生长的稳定期,即细胞生长与产物生成非偶联。在发酵64 h时,肝素前体的产量达到最高,为 (7.25±0.36) g/L,是摇瓶水平的2.51倍,此后产量保持稳定,相比于之前研究中过表达时重组菌在3 L罐上发酵72 h时产量达到最高((5.82±0.17) g/L)[5],本菌株表现出了更高的产量和生产强度。产物的w为 (46.66±2.71) kDa (表3),较摇瓶培养时降低,可能因为搅拌桨机械剪切力的作用使糖链切断,但p值1.28却显示3 L罐发酵仍较好地保持了相对集中的分子量分布。
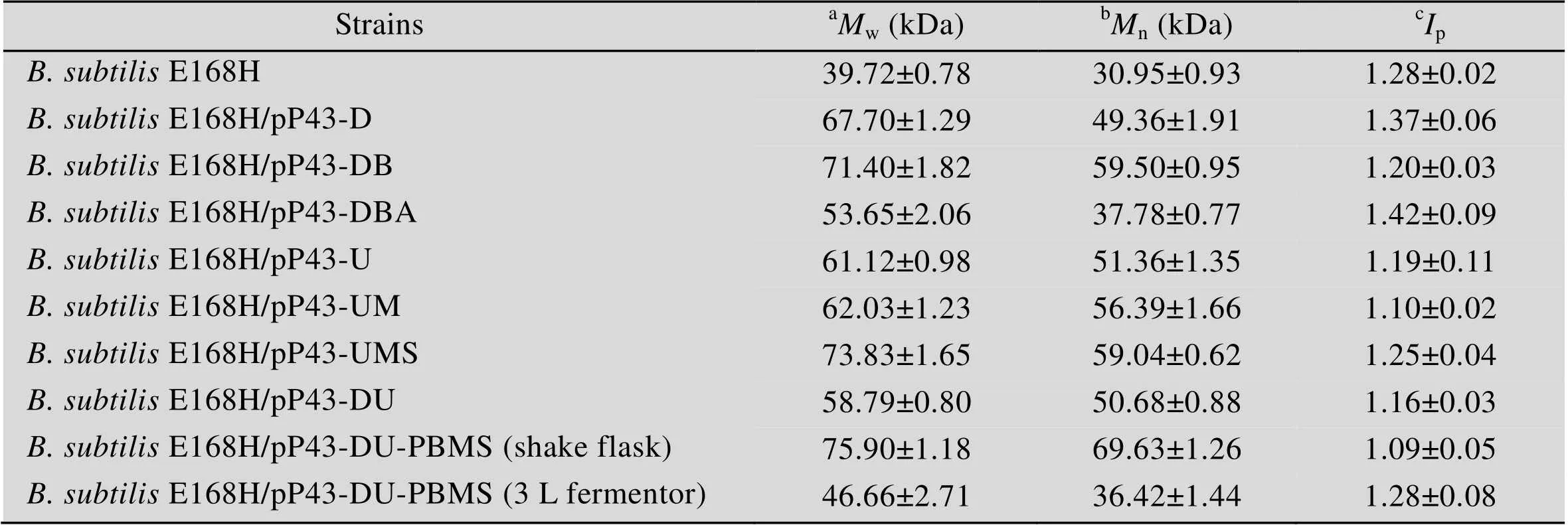
表3 本研究中肝素前体的分子量
aThe weight-average molecular mass (w);bThe number-average molecular mass (w);cPolydispersity index (p).

图4 重组菌B. subtilis E168H/pP43-DU-PBMS在3 L罐中的补料分批培养
3 结论
肝素前体作为一种化学酶法合成肝素的骨架,其产量的提高具有重要意义。在产肝素前体的枯草芽胞杆菌中进行合成途径优化,通过组合过表达UDP-GlcUA途径和UDP-GlcNAc途径中的关键酶基因,构建了8株重组菌。其中,共表达、、、和基因时,肝素前体的产量最高,摇瓶水平为 2.89 g/L,且分子量为75.90 kDa,说明肝素前体的生物合成与分子量调控依赖于两前体物的充足与比例平衡。进一步通过3 L罐补料分批培养,肝素前体最终积累至7.25 g/L,分子量为46.66 kDa。本研究为高产肝素前体提供了新策略,并可将此策略应用于强化其他多糖如透明质酸、软骨素等的生物合成。
[1] Laremore TN, Zhang FM, Dordick JS, et al. Recent progress and applications inglycosaminoglycan and heparin research. Curr Opin Chem Biol, 2009, 13(5/6): 633–640.
[2] Bhaskar U, Sterner E, Hickey AM, et al. Engineering of routes to heparin and related polysaccharides. Appl Microbiol Biotechnol, 2012, 93(1): 1–16.
[3] Liu YF, Liu L, Chen JH, et al. Effects of carbon sources and feeding strategies on heparosan production byK5. Bioprocess Biosyst Eng, 2012, 35(7): 1209–1218.
[4] Liu HY, Zhang ZQ, Linhardt RJ. Lessons learned from the contamination of heparin. Nat Prod Rep, 2009, 26(3): 313–321.
[5] Jin P, Zhang LP, Yuan PH, et al. Efficient biosynthesis of polysaccharides chondroitin and heparosan by metabolically engineered. Carbohydr Polym, 2016, 140: 424–432.
[6] Guerrini M, Beccati D, Shriver Z, et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat Biotechnol, 2008, 26(6): 669–675.
[7] Laurencin CT, Nair L. The FDA and safety—beyond the heparin crisis. NatBiotechnol, 2008, 26(6): 621–623.
[8] Xu YM, Masuko S, Takieddin M, et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science, 2011, 334(6055): 498–501.
[9] Bauer KA, Hawkins DW, Peter PC, et al. Fondaparinux, a synthetic pentasaccharide: the first in a new class of antithrombotic agents—the selective factor Xa inhibitors. Cardiovasc Ther, 2002, 20(1): 37–52.
[10] BoltjeTJ, Buskas T, Boons GJ, et al. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat Chem, 2009, 1(8): 611–622.
[11] Zhang ZQ, McCallum SA, Xie J, et al. Solution structures of chemoenzymatically synthesized heparin and its precursors. J Am Chem Soc, 2008, 130(39): 12998–13007.
[12] Li YH, Yu H, Thon V, et al. Donor substrate promiscuity of the N-acetylglucosaminyltransferase activities ofheparosan synthase 2 (PmHS2) andK5 KfiA. Appl Microbiol Biotechnol, 2014, 98(3): 1127–1134.
[13] Restaino OF, Bhaskar U, Paul P, et al. High cell density cultivation of a recombinantstrain expressing a key enzyme in bioengineered heparin production. Appl Microbiol Biotechnol, 2013, 97(9): 3893–3900.
[14] Wang ZY, Ly M, Zhang FM, et al.K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor. Biotechnol Bioeng, 2010, 107(6): 964–973.
[15] Barreteau H, Richard E, Drouillard S, et al. Production of intracellular heparosan and derived oligosaccharides by lyase expression in metabolically engineeredK-12. Carbohydr Res, 2012, 360: 19–24.
[16] Zhang CY, Liu L, Teng LP, et al. Metabolic engineering ofBL21 for biosynthesis of heparosan, a bioengineered heparin precursor. Metab Eng, 2012, 14(5): 521–527.
[17] Wang ZY, Dordick JS, Linhardt RJ.K5 heparosan fermentation and improvement by genetic engineering. Bioeng Bugs, 2011 2(1): 63–67.
[18] Roman E, Roberts I, Lidholt K, et al. OverexpressionofUDP-glucose dehydrogenaseinresultsin decreased biosynthesis of K5 polysaccharide. Biochem J, 2003, 374(3): 767–772.
[19] Lidholt K, Riesenfeld J, Jacobsson KG, et al. Biosynthesis of heparin. Modulationofpolysaccharidechainlength in acell-freesystem. Biochem J, 1988, 254(2): 571–578.
[20] Bitter T, Muir HM. A modified uronic acid carbazole reaction. Anal Biochem, 1962, 4(4): 330–334.
[21] Widner B, Behr R, Von Dollen S, et al. Hyaluronic acid production in. Appl Environ Microbiol, 2005, 71(7): 3747–3752.
[22] Sugiura N, Baba Y, Kawaguchi Y, et al. Glucuronyltransferase activity of KfiC fromstrain K5 requires association of KfiA: KfiC and KfiA are essential enzymes for production of K5 polysaccharide, N-acetylheparosan. J Biol Chem, 2010, 285(3): 1597–1606.
[23] Hodson N, Griffiths G, Cook N, et al. Identification that KfiA, a protein essential for the biosynthesis of theK5 capsular polysaccharide, is anα-UDP-GlcNAc glycosyltransferase: the formation of a membrane-associated K5 biosynthetic complex requires KfiA, KfiB, and KfiC. J Biol Chem, 2000, 275(35): 27311–27315.
[24] Jin P, Kang Z, Yuan PH, et al. Production of specific-molecular-weight hyaluronan by metabolically engineered168. Metab Eng, 2016, 35: 21–30.
[25] Chen WY, Marcellin E, Steen CA, et al. The role of hyaluronic acid precursor concentrations in molecular weight control in. Mol Biotechnol, 2014, 56(2): 147–156.
[26] Chen WY, Marcellin E, Hung J, et al. Hyaluronan molecular weight is controlled by UDP-N-acetylglucosamine concentration in. J Biol Chem, 2009, 284(27): 18007–18014.
(本文责编 陈宏宇)
Optimization of heparosan synthetic pathway in168
Linpei Zhang1,2, Hao Wang1,2, Zhengxiong Zhou1,2, Guocheng Du1,2, Jian Chen1,2, and Zhen Kang1,2
1,,,214122,,School of BiotechnologyJiangnan UniversityWuxiJiangsuChina
Heparosan is the start point for chemoenzymatic synthesis of heparin and it isof great significance to efficiently synthesize heparosan in microorganisms. The effects of overexpressing key enzyme genes of the UDP-glucuronic acid (UDP-GlcUA) pathway (,and) or the UDP-N-acetyl-glucosamine (UDP-GlcNAc) pathway (,and) on the heparosan production and molecular mass were analyzed in the constructed heparosan-producing((1.71±0.08) g/L). On this basis, heparosan production was increased to (2.89±0.11) g/L with the molecular mass of (75.90±1.18) kDa through co-overexpressing the,,,andgenes in shake flask cultivation. In the 3 L fed-batch fermentation, heparosan production was improved to (7.25±0.36) g/L with the molecular mass of (46.66±2.71) kDa, providing the potential for heparosan industrial production.
heparosan,, pathway optimization, production, molecular mass
10.13345/j.cjb.160453
November 18, 2016; Accepted: December 23, 2016
Zhen Kang. Tel: +86-510-85918307; Fax: +86-510-85918309; E-mail: zkang@jiangnan.edu.cn Guocheng Du. Tel: +86-510-85918307; Fax: +86-510-85918309; E-mail: gcdu@jiangnan.edu.cn
Supported by:National Natural Science Foundation of China (No. 31670092), Key Technologies R & D Program of Jiangsu Province, China (No. BE2014607), Program for Chang Jiang Scholar and Innovative Research Team in University (No. IRT_15R26), Natural Science Foundation of Jiangsu Province (No. BK20141107).
国家自然科学基金(No. 31670092),江苏省科技支撑计划项目(No. BE2014607),长江学者和创新团队发展计划(No. IRT_15R26),江苏省自然科学基金(No. BK20141107 )资助。
猜你喜欢
中国生物防治学报(2022年3期)2022-07-09 10:00:22
天津医科大学学报(2021年1期)2021-12-05 11:11:05
湖南饲料(2021年4期)2021-10-13 07:32:46
微生物学杂志(2021年2期)2021-07-01 11:01:06
微生物学杂志(2020年2期)2020-12-31 07:17:13
扬子江(2019年3期)2019-05-24 14:23:10
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
茶叶通讯(2014年2期)2014-02-27 07:55:40
江汉大学学报(自然科学版)(2013年2期)2013-10-22 07:24:18
河北林业科技(2011年5期)2011-04-10 10:16:21
