
TRAF6与肿瘤关系的研究进展
2017-06-21 12:28李妙周立军
生物技术通报 2017年6期
李妙 周立军
(天津大学药物科学与技术学院 天津市现代药物传递及功能高效化重点实验室,天津 300072)
TRAF6与肿瘤关系的研究进展
李妙 周立军
(天津大学药物科学与技术学院 天津市现代药物传递及功能高效化重点实验室,天津 300072)
肿瘤坏死因子受体相关因子6(Tumor necrosis factor receptor-associated factor 6,TRAF6)是肿瘤坏死因子受体相关因子家族的一员,研究发现其在许多恶性肿瘤中高表达,并且在肿瘤细胞的增殖、迁移和凋亡过程中发挥重要作用。随着对TRAF6与不同类型肿瘤关系的深入研究,干扰或者抑制TRAF6在与肿瘤相关信号通路中的作用或许可以为癌症治疗提供新的策略。根据TRAF6的生物学特性及其在肿瘤相关信号通路中的重要作用,综述了TRAF6与肿瘤的关系,探讨了TRAF6在肿瘤治疗中的重要意义,旨在为今后以TRAF6为靶点的癌症治疗提供理论依据。
肿瘤坏死因子受体相关因子6;信号通路;肿瘤
肿瘤坏死因子受体相关因子(Tumor necrosis factor receptor-associated factors,TRAFs)共包括7个相关蛋白(TRAF1-7),TRAFs最初是在肿瘤坏死因子受体(Tumor necrosis factor receptor,TNFR)介导的信号通路中作为转导分子被发现,之后研究表明:其不仅参与调节受体配体复合物在细胞中的定位,还参与调控关键蛋白在信号通路中的降解与活化[1]。其中,TRAF6作用较独特,因为能同时作为TNFR超家族和Toll样/白细胞介素1受体(Toll/IL-1 receptor,TIR)超家族重要的接头分子[2]。TRAF6在不同细胞系和人体组织中都有表达分布,目前大量的数据提示TRAF6在肿瘤组织中存在异常表达与激活的现象,其中包括食管鳞状细胞癌组织[3]、肺癌组织[4]、结肠癌组织[5]、胰腺癌组织[6]等,这暗示着TRAF6与癌症的发生发展存在着密切关系。本文对TRAF6的生物学特点,其参与调控的主要信号通路,与肿瘤的关系以及其拮抗剂在肿瘤治疗中的研究进展进行了综述,旨在为今后以TRAF6为靶点的癌症靶向治疗提供参考。
1 TRAF6的发现及结构特点
目前哺乳动物中共发现了7种TRAFs蛋白(TRAF1-7),但其人源同系物只有6种(TRAF1-6),其中最早发现的是TRAF1和TRAF2。TRAF6蛋白由530个氨基酸组成,相对分子量为60 kD[7]。最先是由Ishida等[7]于1996年利用酵母双杂交方法对CD40信号转导进行研究时发现的。TRAF6基因从果蝇到人类都非常保守。同其他TRAFs一样,TRAF6 C端含有一个高度保守的TRAF结合结构域,X射线晶体学研究发现TRAF结构域呈三聚蘑菇头状结构,该结构域又被划分为CC卷曲螺管样结构域(茎)和TRAF-C(头)两部分[8],能够分别同受体家族胞内段的TRAF结构域或胞浆中其他含有TRAF结构域的蛋白结合形成二聚体或者三聚体。TRAF6 N端含有一个保守的RING环指结构域(67-124位)和5个锌指结构域(130-236位)。RING环指结构域及第一个Zn指结构域是TRAF6作为E3泛素连接酶所必须的,是目前发现的TRAF6蛋白主要功能区域,与E2泛素结合酶Ubc13/Uev1A结合后可以共同调节包括自身在内的多种蛋白的多聚泛素化,并负责下游信号通路的激活(图1)[9]。
2 TRAF6信号转导及调节机制
2.1 TRAF6信号转导
在哺乳动物中,TRAF6不仅同其他家族成员TRAF2/3/5的结构具有一定的相似性,而且也参与了这些蛋白所调控的一系列信号通路[10]。然而因其独特的TRAF-C结构域,TRAF6所识别的底物分子与其他成员所识别的底物分子相比具有明显的结构差异。晶体结构分析表明:底物分子与TRAF6的结合位点为X-X-P-X-E-X-X-芳香/酸性序列[8],而TRAF2/3/5识别的却是其他形式的序列,如P-XQ-X-T[11,12]。虽然结合序列存在差异性,但TRAF6却可以被招募至其他蛋白复合物上,其中便包括TRAFs蛋白复合物,如一些炎症信号通路就是在TRAF6与TRAF3形成复合体后才被激活的[13]。复合物的存在行式能够介导TRAF6与受体发生作用。一种方式是直接介导TRAF6与膜受体胞内端结合,如TNFR超家族[8,11]和TGFβRI/ALK5[14,15]。另一种方式是通过胞质中的中介蛋白间接介导TRAF6与受体结合,如利用IRAK1激酶与TLR/IL-1R超家族结合[8],利用MALT1与T细胞受体(TCR)结合[16]。TRAF6信号复合物不仅含有能与TRAF6结合的蛋白,还包含一些对于募集激酶及其他配体蛋白所必需的信号元件。最值得关注的是TLR/IL-1R信号途径,TRAF6同时介导了髓样分化因子88(MyD88)依赖和非依赖的信号途径:TLR/IL-R(TLR4,TLR9和IL-1R)受到信号刺激后,TRAF6会被招募到Myddosome复合体,该复合物包含配体蛋白MyD88和IRAK1/2/4[17];TLR3受到信号刺激后,TRAF6将被招募到另一个复合物,该复合物包含配体蛋白TRIF,家族成员TRAF3以及RIP1激酶,伴随着TRAF6的活化TAK1也会被激活[13,18,19]。TRAF6自身还可与受体胞内区直接结合,之后再形成蛋白复合物。例如,TRAF6与TNFR超家族成员直接结合后,再利用一些支架蛋白(c-Cbl,Cbl-b以及RACK1)招募下游激酶(c-Src和PI3K)(图1)[20-22]。
2.2 TRAF6的信号通路及其调节机制
TRAF6的胞内调节机制可以分为两种:一种是针对其泛素连接酶活性的去泛素化调节;另一种是通过交叉作用机制进行调节。与其他的翻译后修饰类似,泛素化修饰是一个可逆的过程。如今,CYLD和A20都被证实能够负调控TRAF6介导的泛素化修饰[23,24]。肿瘤抑制因子CYLD被接头蛋白p62招募到TRAF6后,通过自身去泛素酶活性阻止了TRAF6的泛素化,从而负调控RANK信号通路[25]。因此,CYLD功能异常可能导致TRAF6过度活化,从而引起一系列病理反应。具有潜在肿瘤抑制作用的A20同样可以通过一系列机制负调控TRAF6信号通路,除了通过触发经典的Lys-48多聚泛素化介导Ubc13的降解来阻止TRAF6复合物的泛素化修饰[26],还可以通过移除TRAF6催化连接的Lys-63泛素链来终止TLR/IL-1R的信号转导过程[24]。
最近越来越多的研究表明TRAF6与其他通路的交叉作用也可调节其在通路中信号传递(图1)。虽然大多数研究聚焦于典型的TRAF6信号机制,即TRAF6-TAK1-MAPK/IKK-NF-κB,但是TRAF6也参与了许多其他信号转导过程,如TRAF6也能够激活PI3K及下游AKT/PKB信号通路。JAK-STAT信号通路通常被认为是与TRAF6介导的信号通路相互独立的,但一些证据表明这两条信号通路同样存在交叉。STAT3是JAK-STAT通路中的一种转录因子,在多种肿瘤细胞和组织中均有过度表达,持续激活的STAT3为肿瘤形成和发展所必需。研究发现,TRAF6在不改变STAT3蛋白表达量的基础上能够促进STAT3的Lys-63泛素化,进一步激活STAT3转录因子的靶基因,如MCL1和Bcl-2[27,28]。然而,STAT3通过下调Ubc13的表达也可以负反馈调节TRAF6信号通路[29]。另外,TRAF6与Notch信号通路也存在交叉。首先,伴随Notch配体表达增加,缺乏TRAF6的骨骼肌卫星细胞增殖仍呈上升趋势[30];其次,与TRAF6激活相关的TGFβ受体可以招募早老素1(PS1)至TGFβRI复合物并促使TGFβRI剪切入核,PS1同样能够促进Notch受体胞内段剪切入核[31]。以上研究间接表明两者之间存在交叉调控。Notch信号通路在进化上高度保守,负责调控细胞的发育和分化等多个过程。Notch信号通路在肿瘤细胞中存在过度激活,这使得对TRAF6在Notch信号通路中的作用的研究成为一个重要的新方向,或许可以用来解释TRAF6是如何调节炎症反应,以及如何巧妙的控制各种肿瘤干细胞的分化。
TRAF6作为泛素连接酶催化目的蛋白发生Lys-63泛素化修饰进而激活一系列信号通路(图1)。TRAF6与E2泛素结合酶Ubc13/Uev1A结合后,通过泛素化AKT的K8与K14位点,能够促进AKT向细胞膜的移位,从而参与调控PI3K/AKT信号通路[32]。活化的AKT可通过磷酸化作用激活或抑制其下游靶蛋白产生促进癌细胞的增殖与抗凋亡效果[33]。TRAF6首先与膜上接头蛋白TAB2结合,之后形成TRAF6/TAK1/TAB1/TAB2复合物,复合物与Ubc13/Uev1A的绑定将激活TAK1。活化的TAK1可以分别激活下游的IKK复合物和MAPK途径。有研究发现,含RING锌指结构域的TRAF6可以介导IKK调节亚基NEMO(IKKγ)发生Lys-63泛素化[34,35]。另外,NEMO泛素化将会引起IKK复合体的激活,使NF-κB的胞浆抑制因子IκB磷酸化降解并促使NF-κB的核定位序列暴露出来迅速转位入核,在核内与相关靶基因特异序列结合启动核基因转录。TRAF6还可以通过TAK1依赖方式诱导JNK与p38的活化,导致转录因子AP-1家族成员jun和fos活化促进炎症因子产生,或者通过线粒体途径调节半胱氨酸天冬氨酸特异性蛋白酶(Caspase)的活性来调控癌细胞的凋亡[36]。除了通过TRAF6-TAK1-JNK/p38激活MAPK信号转导过程,还可经由TRAF6-Ras-ERK激活MAPK信号通路。研究表明TNF受体家族成员跨膜蛋白CD40受到信号刺激后,TRAF6则将会与CD40胞质部分结合介导下游Ras激酶活化,从而引起三级酶联反应激活ERKs信号通路,活化的ERKs负向调控癌细胞的增殖、分化与凋亡等[37,38]。除了上述提到的TAK1和AKT,TRAF6也可通过改变活性氧(ROS)水平间接增强NF-κB活化。NF-κB活化可以促进DNA复制,G1/S期转录,上调CyclinD1的表达从而使细胞过度增殖导致癌症的发生;还可抑制caspase3激活达到抗凋亡的目的;也可促进炎症因子产生为肿瘤细胞生存提供微环境[39]。此外,TRAF6也能够激活Src家族非受体酪氨酸激酶,如c-Src[22],该家族成员都是由原癌基因编码参与细胞内多条信号途径。TRAF6还可以介导缺氧诱导因子-1α(HIF-1α)发生以Lys-63连接的多聚泛素化,使其稳定性提高并且这种调节作用不依赖于氧气。HIF-1α是转录因子,调节多种基因的表达,在肿瘤血管生成和肿瘤发展过程中发挥非常重要的作用[40]。上述TRAF6所介导的信号通路都与肿瘤发生发展具有密切关系,所以也表明TRAF6在调控与肿瘤相关的信号过程中具有重要作用。
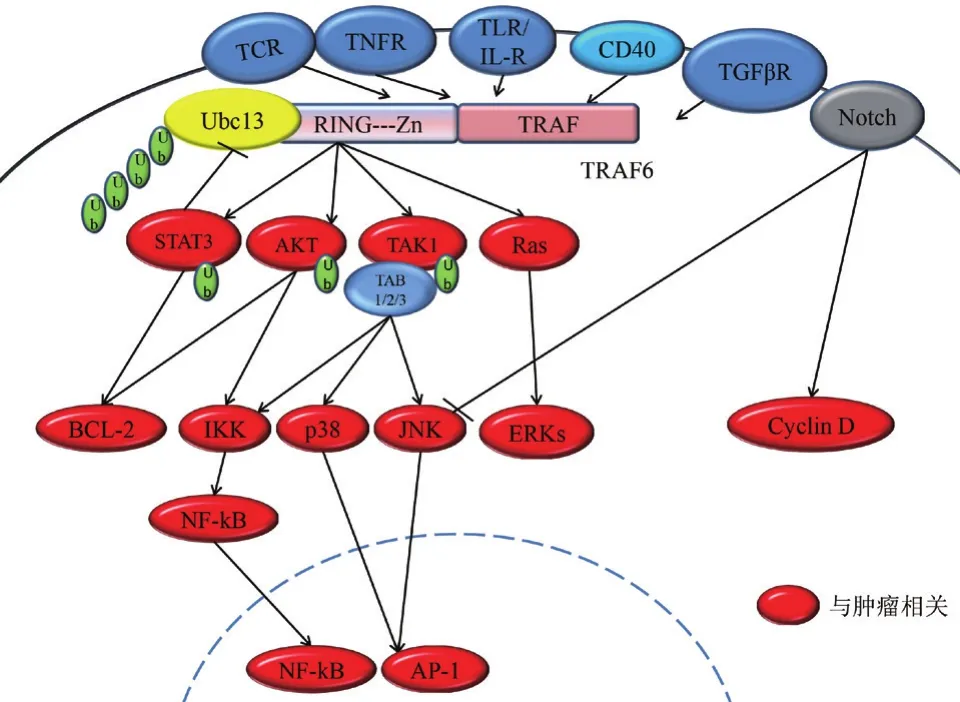
图1 TRAF6与肿瘤相关信号通路图[8-40]
3 TRAF6与肿瘤的关系
3.1 TRAF6与肿瘤的研究进展
细胞恶性增殖、凋亡失衡、发生侵袭和转移等是肿瘤细胞恶性生物学行为,大部分癌症病人最后死于癌症的复发与转移。大量的实验表明TRAF6几乎在所有有关报道过的肿瘤细胞系中都存在过表达,并具有调控肿瘤细胞恶性行为的功能。首先,研究表明过表达的TRAF6诱导了肿瘤发生,以至于一部分科学家认为TRAF6为原癌基因。例如,有研究证明胶质母细胞瘤和头颈部癌症的发病机制是discoidina神经毡状膜蛋白通过上调TRAF6,促进AKT磷酸化,从而诱导了肿瘤发生[41]。动物实验显示抑制TRAF6的表达可以明显阻止肿瘤生长和肿瘤血管的生成[40]。其次,致癌基因Ras是TRAF6调控肿瘤细胞生物学行为的重要下游靶点,目前已知TRAF6通过影响Ras激酶活性不仅增强了癌细胞的侵袭性还提高了癌细胞的转移性。Yao等[3]发现无论是体内实验还是体外实验敲除TRAF6基因都会阻止食管鳞癌细胞的转移,并对其转移机制进行了研究,证明是TRAF6的N端与Ras接合导致ERK磷酸化的结果。Starczynowski等[42]发现在非小细胞型肺癌(NSCLC)与小细胞型肺癌(SCLC)中TRAF6在基因层次上都相对上调,过表达的TRAF6引发依赖Ras通路的肿瘤生成。此外,过表达TRAF6不仅能够抑制各种类型癌细胞的凋亡还可促进癌细胞增殖。Yao等[3]应用RT-PCR与免疫组化法均证实TRAF6在食管癌组织中表达升高,为了进一步探讨两者的相关性,他们利用siRNA下调了Eca109细胞中TRAF6的表达,发现细胞生长受到抑制、致瘤性显著降低、细胞凋亡比例增高,与细胞凋亡相关的蛋白caspase3、PARP等均发生相应的改变。在SPC-A-1肺腺癌细胞中,下调TRAF6基因的表达会抑制癌细胞迁移、侵袭甚至会促进细胞凋亡,进一步探讨发现这可能是由于TRAF6参与调控NF-κB-CD24/CXCR4信号通路的结果[43]。Sun等[5]发现,当用siRNA沉默人结肠癌细胞中TRAF6基因后,细胞增殖减慢,细胞G1期比例显著增高,调控G1期向S期过渡的重要因子CyclinD1在体内体外的表达均下降,由此推断TRAF6或许是通过参与细胞分裂帮助结肠癌细胞增殖。总之,TRAF6在异常表达的基础上利用自身酶活性调节下游靶蛋白的活性,从而参与调控癌细胞恶性生物学行为。虽然目前TRAF6异常表达的机制研究还不够透彻,但是我们推测可能是由于癌症患者的体内环境发生了变化,免疫系统便试图消灭体内过度增殖的癌细胞。
3.2 靶向TRAF6结构域治疗策略
3.2.1 完全抑制TRAF6 目前大量数据提示,TRAF6在恶性肿瘤中高表达,控制其表达可以达到抑制肿瘤生长的作用,然而值得注意的是TRAF6在天然免疫和获得性免疫中同样发挥重要作用。免疫细胞表面受体活化时TRAF6被直接或者间接募集到受体的胞内结构域,激活一系列信号级联反应,从而诱导免疫和炎症反应。研究发现,TRAF6-/-小鼠的脾、胸腺萎缩,最终会死于重症石骨症[44,45]。而且TRAF6缺失会造成CD5+B1细胞生成受限,使得骨髓和脾脏中的成熟B细胞数量减少,并导致T细胞依赖性和非依赖性的体液免疫反应[46]。此外,TRAF6还是树突状细胞成熟和活化所必需的,TRAF6-/-树突状细胞不能有效上调相关炎症细胞因子的产生。综上所述,如果完全抑制TRAF6表达将使得机体免疫功能发生紊乱。所以,以TRAF6为靶点的抑制剂研究也要考虑在免疫方面带来的副反应。由于TRAF6结构的独特性使其功能也具有多样性,目前研究聚焦于靶向针对其特定的结构域,以期既达到良好的抗肿瘤效果,又能够正确调控它在免疫反应中的作用。
3.2.2 抑制TRAF6部分结构域 抑制C端结合结构域:TRAF6在免疫方面主要依靠CD40受体发挥作用。有研究人员发现TRAF6(333-508位)通过自身结合肽与CD40结合,进而调控下游与免疫相关的信号通路[8]。然而在TRAF6缺陷细胞中,两种TRAF6突变体(缺失357-530位;TRAF6T471A突变)虽然都不能与CD40绑定,但是依然能够恢复CD40依赖的JNK活化和CD80的上调[47]。同样也有研究表明,突变CD40中与TRAF6的结合位点(缺失231-246位;232位与235位谷氨酸被丙氨酸取代)也不会破坏其激活JNK,NF-κB或CD80上调的能力[48-50],以上这些数据不仅表明存在着CD40-TRAF6的代偿机制而且进一步表明TRAF6 C端结合结构域可以通过直接或者间接方式参与上述过程。利用siRNA技术沉默TRAF6 C端结构域能够有效抑制黑色素瘤的生长[51],这可能是由于干扰了与TRAF6相关的下游通路。例如,有研究证明诱导性热休克蛋白70(Heat shockprotein 70,HSP70)与TRAF6的C端结构域结合将阻止TRAF6的泛素化继而抑制了NF-κB活化[52]。另有研究发现siRNA TRAF6 C端结构域(420-440位)只会降低TRAF6蛋白的表达而不影响其与受体低聚化,最终也能显著降低小鼠骨髓瘤细胞数目。因此,靶向抑制TRAF6 C端结构域或许能够达到既不影响免疫功能又能够有效遏制癌细胞生长的目的。
抑制N端功能结构域:通过TRAF6 siRNA沉默特定Zn指结构域(233-253位)后,TRAF6蛋白表达量基本上等同于未沉默组[53],并没有对小鼠骨髓瘤细胞的生长与增殖产生影响,但是对于免疫方面的影响并未有过报道。相反,无环TRAF6突变体将导致CD40信号途径异常,使得CD40介导B细胞分泌抗体的过程受阻并出现多种与免疫相关的病理反应[54]。已知C70残基位于TRAF6-Ubc13复合物的结合位点上(即RING环指结构域),Yang等[55]发现C70A突变的TRAF6不具有泛素连接酶活性,以至于AKT的蛋白功能受限,已知AKT在癌症的发生发展中同样扮演着重要角色。相比于TRAF6的C端结构域,TRAF6的RING环指结构域在其所介导的CD40信号通路中表现出了不可或缺的必要性。因此,直接干扰RING环指结构域虽然能够抗肿瘤,但会伴随免疫低下。然而,有研究组成功构建出Ubc13+/-小鼠模型后发现虽然Ubc13蛋白表达水平显著降低,但小鼠免疫系统仍然正常。不仅如此,Ubc13+/-小鼠与野生型同窝小鼠相比并没有增加感染、自身免疫和恶性肿瘤的发病率[56]。由此可见,控制E2结合酶Ubc13或许并不会带来对免疫系统的副作用,也间接提示在不破坏TRAF6自身RING环指结构域的基础上阻止其发挥E3泛素连接酶活性或许并不会影响机体正常的免疫功能。
3.3 靶向TRAF6拮抗剂的研究
TRAF6在癌症的病理机制中发挥重要作用,或许可以作为癌症治疗的靶标。一方面,TRAF6在高表达的基础上通过自身酶活性调控了肿瘤细胞的恶性生物学行为;另一方面,大部分恶性肿瘤都存在局部炎症,当机体受到炎症侵袭时也会激活TRAF6,这对于炎症因子的释放起着重要调控作用,相对高表达的TRAF6会利用自身活性促进适宜癌细胞生存的炎性微环境的形成。因此针对TRAF6进行拮抗物相关的药物设计意义重大。目前研究中最为常用的TRAF6抑制剂是MG132,MG132是一种有效、可逆的醛基肽类26S蛋白酶体抑制剂,能够抑制TRAF6的表达[57]。近年,TRAF6抑制剂已经被尝试用于促进癌细胞凋亡的研究。最初,人们发现20S蛋白酶体抑制Bortezomib具有抑制骨髓瘤细胞增殖的作用,进一步研究发现:Bortezomib可以抑制TRAF6泛素化从而引起其下游信号通路的改变,使得骨髓瘤细胞增殖减慢[58]。不久,特异性针对TRAF6活性研究的小分子化合物也被发现,如小白菊内酯[59],研究发现该化合物能够有效抑制TRAF6活性,并显著降低TRAF6下游NEMO的泛素化,最终有效诱导了骨髓瘤细胞的凋亡。我们的最新研究也发现,喹啉类化合物奎宁通过阻碍TRAF6对AKT的激活作用,通过诱导癌细胞凋亡来抑制癌细胞增殖[60]。首先,抑制TRAF6活性可以阻碍癌细胞增殖促进癌细胞凋亡这一观点得到了实验的支持;其次,虽然TRAF6的异常表达机制研究还不够透彻,但是有足够研究表明,直接干扰TRAF6的表达也可以大大减缓癌细胞的增殖[61,62]。此外,靶向抑制TRAF6对免疫方面的影响仍需进一步研究,从而使靶向TRAF6抑制剂在肿瘤微环境中更好的发挥作用。这些研究将使得抑制或者干扰TRAF6为靶点的治疗成为一个令人关注的治疗策略。
4 展望
一个世纪以来,除了心血管类疾病,癌症逐渐发展成为威胁人类健康的杀手。TRAF6的过表达与肿瘤的发生发展密切相关,通过介导不同的信号通路,参与调控肿瘤细胞凋亡,生长和侵袭等,对以TRAF6为靶点的抗癌作用机制的深入研究可以为临床肿瘤的靶向治疗提供更多的基础理论依据,为新的抗癌药物的发现提供新的思路。
[1]Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors(TRAFs)[J]. Oncogene, 2001, 20(44):6482-6491.
[2]Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling[J]. Curr Protoc Immunol, 2009, doi:10.1002/0471142735. im1109ds87.
[3]Yao F, Han Q, Zhong C, et al. TRAF6 promoted the tumorigenicity of esophageal squamous cell carcinoma[J]. Tumor Biology, 2013, 34(5):3201-3207.
[4]Zhang XL, Dang YW, Li P, et al. Expression of tumor necrosis factor receptor-associated factor 6 in lung cancer tissues[J]. Asian Pac J Cancer Prev, 2014, 15(24):10591-10596.
[5]Sun H, Li X, Fan L, et al. TRAF6 is upregulated in colon cancer and promotes proliferation of colon cancer cells[J]. Iubmb Life, 2012, 53(9):775-782.
[6]Rong Y, Wang D, Wu W, et al. TRAF6 is over-expressed in pancreatic cancer and promotes the tumorigenicity of pancreatic cancer cells[J]. Medical Oncology, 2014, 31(11):1-10.
[7]Ishida T, Mizushima S, Azuma S, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region[J]. J Biol Chem, 1996, 271(46):28745-28748.
[8]Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology[J]. BioEssays, 2003, 25(11):1096-1105.
[9]Lamothe B, Campos AD, Webster WK, et al. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL[J]. J Biol Chem, 2008, 283(36):24871-24880.
[10]Xie P. TRAF molecules in cell signaling and in human diseases[J]. J Mol Signal, 2013, 8(1):1-31.
[11]Chung JY, Lu M, Yin Q, et al. Structural revelations of TRAF2 function in TNF receptor signaling pathway[J]. Advances in Experimental Medicine and Biology, 2006, 597(4):93-113.
[12]Hildebrand JM, Luo Z, Manske MK, et al. A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling[J]. J Exp Med, 2011, 207(12):2569-2579.
[13]Hacker H, Redecke V, Blagoev B, et al. Specificity in toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6[J]. Nature, 2006, 439(7073):204-207.
[14]Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinaseindependent manner[J]. Nat Cell Biol, 2008, 10(10):1199-1207.
[15]Yamashita M, Fatyol K, Jin C, et al. TRAF6 mediates Smadindependent activation of JNK and p38 by TGF-beta[J]. Mol Cell, 2008, 31(6):918-924.
[16]Sun L, Deng L, Ea CK, et al. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes[J]. Mol Cell, 2004, 14(3):289-301.
[17]Cohen P. The TLR and IL-1 signalling network at a glance[J]. J Cell Sci, 2014, 127(11):2383-2390.
[18]Cussonhermance N, Khurana S, Lee TH, et al. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-kB activation but does not contribute to interferon regulatory factor 3 activation[J]. J Biol Chem, 2005, 280(44):36560-36566.
[19]Kawasaki T, Kawai T. Toll-like receptor signaling pathways[J]. Front Immunol, 2014, 5:461.
[20]Arron JR, Vologodskaia M, Wong BR, et al. A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine(trance)and CD40L-mediated Akt activation[J]. J Biol Chem, 2001, 276(32):30011-30017.
[21]Neumann D, Lienenklaus S, Rosati O, et al. IL-1beta-induced phosphorylation of PKB/Akt depends on the presence of IRAK-1[J]. Eur J Immunol, 2002, 32(12):3689-3698.
[22]Wong BR, Besser D, Kim N, et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src[J]. Mol Cell, 2000, 4(6):1041-1049.
[23]Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses[J]. Nat Immunol, 2004, 5(10):1052-1060.
[24] Trompouki E, Hatzivassiliou E, Tsichritzis T, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members[J]. Nature, 2003, 424(6950):793-796.
[25]Jin W, Chang M, Paul EM, et al. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice[J]. J Clin Invest, 2008, 118(5):1858-1866.
[26]Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes[J]. Science, 2010, 327(5969):1135-1139.
[27]Yoshida Y, Kumar A, Koyama Y, et al. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism[J]. J Biol Chem, 2004, 279(3):1768-1776.
[28]Wei J, Yuan Y, Jin C, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination[J]. PLoS One, 2012, 7(11):e49567.
[29]Zhang H, Hu H, Greeley N, et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13[J]. Nat Commun, 2013, 5:5798-5798.
[30]Hindi SM, Paul PK, Saurabh D, et al. Reciprocal interaction between TRAF6 and notch signaling regulates adult myofiber regeneration upon injury[J]. Mol Cell Biol, 2012, 32(23):4833-4845.
[31]Gudey SK, Sundar R, Mu Y, et al. TRAF6 stimulates the tumor-promoting effects of TGFbeta type I receptor through polyubiquitination and activation of presenilin 1[J]. Sci Signal, 2014, 7(307):ra2.
[32]邱文, 单锴, 庞蓉蓉, 等. 大鼠野生型TRAF6基因和TRAF6 shRNA表达质粒的构建及鉴定[J]. 南京医科大学学报:自然科学版, 2011, 31(10):1407-1411.
[33]Shinohara M, Yun JC, Saji M, et al. Minireview:AKT in thyroid tumorigenesis and progression[J]. Endocrinology, 2007, 148(3):942-947.
[34]王敏, 王晓辉, 唐刘君, 等. TRAF6截短体的构建及其对NF-κB信号通路的影响[J]. 安徽医科大学学报, 2011, 46(11):1113-1116.
[35]Lamothe B, Campos AD, Webster WK, et al. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL[J]. J Biol Chem, 2008, 283(36):24871-24880.
[36]Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development[J]. Nat Rev Cancer, 2009, 9(8):537-549.
[37]Heidelberg SB. Tumor Necrosis Factor Receptor-Associated Factor[M]. Berlin Heidelberg:Springer, 2005:1930.
[38]Kashiwada M, Shirakata Y, Inoue JI, et al. Tumor necrosis factor receptor-associated factor 6(TRAF6)stimulates extracellular signal-regulated kinase(ERK)activity in CD40 signaling along a ras-independent pathway[J]. J Exp Med, 1998, 187(2):237-244.
[39]Perkins ND. NF-κB:tumor promoter or suppressor[J]. Trends Cell Biol, 2004, 14(2):64-69.
[40]Sun H, Li XB, Meng Y, et al. TRAF6 upregulates expression of HIF-1α and promotes tumor angiogenesis[J]. Cancer Research, 2013, 73(15):4950-4959.
[41]Feng H, Lopez GY, Kim CK, et al. EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis[J]. J Clin Invest, 2014, 124(9):3741-3756.
[42]Starczynowski DT, Lockwood WW, Deléhouzée S, et al. TRAF6 is an amplified oncogene bridging the RAS and NF-kB pathways in human lung cancer[J]. J Clin Invest, 2011, 121(10):4095-4105.
[43] 林根, 黄传钟, 苏光建, 等. 下调TRAF6表达对肺癌细胞株恶性生物学行为的影响[J]. 中国肺癌杂志, 2015, 18(11):661-667.
[44]Lomaga MA, Yeh WC, Sarosi I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling[J]. Genes Dev, 1999, 13(8):1015-1024.
[45]Naito A, Azuma S, Tanaka S, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice[J]. Genes to Cells, 1999, 4(6):353-362.
[46]Kobayashi T, Kim TS, Jacob A, et al. TRAF6 is required for generation of the B-1a B cell compartment as well as T celldependent and-independent humoral immune responses[J]. Plos One, 2009, 4(3):285-289.
[47]Rowland SL, Tremblay MM, Ellison JM, et al. A novel mechanism for TNFR-associated factor 6-dependent CD40 signaling[J]. J Immunol, 2007, 179(7):4645-4653.
[48]Hostager BS, Haxhinasto SA, Rowland SL, et al. TRAF2-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling[J]. J Biol Chem, 2003, 278(46):45382-45390.
[49]Ahonen CL, Manning EM, Erickson LD, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells[J]. Nature Immunology, 2002, 3(5):451-456.
[50]Jabara H, Laouini D, Tsitsikov E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching[J]. Immunity, 2002, 17(3):265-276.
[51]Liu H, Tamashiro S, Baritaki S, et al. TRAF6 activation in multiple myeloma:a potential therapeutic target[J]. Clinical Lymphoma Myeloma and Leukemia, 2012, 12(12):155-163.
[52]Chen H, Wu Y, Zhang Y, et al. Hsp70 inhibits lipoplysaccharideinduced NF-Kb activation by interaction with TRAF6 and inhibiting its ubiquitination[J]. Febs Letters, 2006, 580(13):3145-3152.
[53]Chen H, Li M, Campbell RA, et al. Interference with nuclear factor kappa B and c-Jun NH2-terminal kinase signaling by TRAF6C small interfering RNA inhibits myeloma cell proliferation and enhances apoptosis[J]. Oncogene, 2006, 25(49):6520-6527.
[54]Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6(TRAF6)in CD40-mediated B lymphocyte effector functions[J]. J Immunol, 2000, 164(2):623-630.
[55]Yang WL, Wang J, Chan CH, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation[J]. Science, 2009, 325(5944):1134-1138.
[56]Fukushima T, Matsuzawa S, Kress CL, et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor(TRAF)-mediated inflammatory responses[J]. Proceedings of the National Academy of Sciences, 2007, 104(15):6371-6376.
[57]Xu G, Wen H, Zhou H, et al. Involvement of IRAKs and TRAFs in anti-beta(2)GPI/beta(2)GPI-induced tissue factor expression in THP-1 cells[J]. Thromb & Haemost, 2011, 106(6):1158-1169.
[58]Aadams J, Kauffman M. Development of the proteasome inhibitor Velcade(Bortezomib)[J]. Cancer Invest, 2004, 22(2):304-311.
[59]Kong FC, Zhang JQ, Zeng C, et al. Inhibitory effects of parthenolide on the activity of NF-κB in multiple myeloma via targeting TRAF6[J]. Journal of Huazhong University of Science & Technology, 2015, 35(3):343-349.
[60]Liu W, Qi Y, Liu L, et al. Suppression of tumor cell proliferation by quinine via the inhibition of the tumor necrosis factor receptor-associated factor 6-AKT interaction[J]. Mol Med Rep, 2016, 14(3)2171-2179.
[61]Hou J, Wang P, Lin L, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2[J]. J Immunol, 2009, 183(3):2150-2158.
[62]Hurst DR, Edmonds MD, Scott GK, et al. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis[J]. Cancer Res, 2011, 69(69):1279-1283.
(责任编辑 狄艳红)
Research Progress on the Relationship Between TRAF6 and Tumor
LI Miao ZHOU Li-jun
(Tianjin Key Laboratory for Modern Drug Delivery & High-Efficiency,School of Pharmaceutical Science and Technology,Tianjin University,Tianjin 300072)
Tumor necrosis factor receptor-associated factor 6(TRAF6)is a member of the super TRAF family. It is not only overexpressed in many cancer tissues but also closely associated with tumor cell proliferation,migration,and apoptosis. Studies on relationships between TRAF6 and different types of tumors have emerged recently,and these investigations suggest that interfering or depressing TRAF6 in the role of tumor-related signaling pathways may provide a new therapeutic approach for the treatment of cancers. This review summarizes the correlation between TRAF6 and tumor based on the biological functions of TRAF6 and its critical roles in tumor-related signaling pathways,and explores the significance of TRAF6 in combatting cancers,aiming at providing theoretical basis for the treatment of cancer by targeting TRAF6 in the future.
tumor necrosis factor receptor-associated factor 6;signaling pathway;tumor
10.13560/j.cnki.biotech.bull.1985.2016-1137
2016-12-18
国家自然科学基金项目(81641132)
李妙,女,硕士研究生,研究方向:抗肿瘤;E-mail:963834485@qq.com
周立军,女,副教授,研究方向:恶性肿瘤;E-mail:lijunzhou@tju.edu.cn
猜你喜欢
奥秘(创新大赛)(2019年9期)2019-10-09
广州大学学报(自然科学版)(2019年1期)2019-05-07
小哥白尼(趣味科学)(2019年1期)2019-04-12
奥秘(2017年5期)2017-07-05
天津科技大学学报(2016年1期)2016-02-28
环境与生活(2016年6期)2016-02-27
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
