
加速溶剂萃取法与索氏提取法对废线路板中二噁英测定的影响
2017-06-21 13:56蔡璐赵文杰周全法
分析化学 2017年5期
蔡璐+赵文杰+周全法


摘 要 将经过多级破碎分选所得的废线路板粉末过60目筛,以2 mol/L HCl溶液、超纯水洗涤,并用丙酮脱水。将预处理后的滤渣、滤纸填入萃取池,或者用新滤纸包裹后置于索氏提取管内,添加5 μL内标物,分别用加速溶剂萃取法(ASE)或索氏提取(SE)法进行萃取,依次采用多层硅胶柱和活性炭柱对萃取提取物进行净化、洗脱,得到二噁英测试样品。用同位素稀释高分辨气相色谱质谱联用仪分析样品中二噁英的含量。考察两种提取方法及不同氯代数对15种13C2,3,7,8PCDD/Fs回收率的影响,比较两种方法的准确度和精密度。结果表明,ASE和SE对15种13C2,3,7,8PCDD/Fs的回收率分别为54.3%~113.0%和28.3%~77.7%, 实测废线路板中二噁英毒性当量(Toxic equivalent quangtity,TEQ)分别为0.075 ng TEQ/kg和0.266 ng TEQ/kg。在方法精密度允许范围内,ASE相对具有简单快速、溶剂用量少和准确的优势。
关键词 二噁英; 索氏提取; 快速溶剂萃取; 废线路板
1 引 言
电器电子产品相关废弃物,如废线路板,处置过程释放二噁英等持久性有机污染物(POPs),对生态环境和人类健康造成了严重危害[1,2]。二噁英类POPs包括多氯代二苯并对二噁英(Polychlorinated Dibenzopdioxins,PCDDs)和多氯代二苯并呋喃(Polychlorinated dibenzofurans,PCDFs),具有高毒性、致癌性和致突变性[1],特别是2,3,7,8位氯取代同系物的毒性更强,危害更严重[3,4]。
对电子废弃物中二噁英类POPs进行有效减控的前提是必须准确测定处理前后物料中二噁英类POPs的含量。目前,对环境空气样品[1,5]、垃圾焚烧飞灰样品[6]和肉类食品[7,8]中2,3,7,8PCDD/Fs的分析檢测研究较多,但对检测和控制电子废弃物处理处置过程中产生的二噁英类POPs的报道较少。二噁英类物质一般以超痕量状态存在,分析检测过程对选择性和灵敏度要求很高,因此必须进行预处理富集。加速溶剂萃取(Accelated solvent extraction, ASE)和索氏提取(Soxhelt extraction,SE)是常用的二噁英类POPs的富集方法[4,9,10]。SE是传统的富集方式,具有仪器设备要求低和操作简便的特点,而ASE则具有快速、高效、自动化、节省溶剂等优点,已被美国EPA 1613标准采用[11]。目前,很多研究采用加速溶剂萃取法提取土壤[12]、奶粉[13]、沉积物[14]等固体样品中的二噁英。Brockmeyer等[15]采用加速溶剂萃取法对固体样品中有机污染物的提取更高效、快捷。Zhang等[16]对比加速溶剂萃取(ASE)、微波提取(MAE)、超声波提取(UAE)这3种萃取方法对有机污染物(PCBs, HCHs, DDts)的提取效果,研究表明,ASE具有更好的提取效果。
本研究分别采用ASE和SE富集,高分辨气相色谱(High resolution gas chromatography,HRGC)高分辨质谱(High resolution magnetic sector,HRMS)联用,测定15种内标物(13C2,3,7,8PCDD/Fs,13CLabeled2,3,7,8Polychlorinated Dibenzopdioxins/furans)的回收率,考察和比较ASE和SE两种方法的准确度及精密度,并对废线路板中的二噁英进行定性和定量分析。
2 实验部分
2.1 仪器与试剂
Trace GC Ultra高分辨气相色谱仪、TRDIOXIN5MS毛细管柱(60 m×0.25 mm×0.25 μm)、DFS高分辨质谱仪(美国Thermo公司); ASE 350加速溶剂萃取仪(美国戴安公司); EYELAN1200B旋转蒸发仪(日本东京理化公司); Simplicity超纯水机(美国密理博公司); HSC12B氮吹仪(天津恒奥公司)。
甲苯、甲醇、正己烷、二氯甲烷、壬烷(农残级,TEDIA公司); 硅胶(100~200目,柱层析用,国药集团); 无水Na2SO4(分析纯,国药集团); HCl、浓H2SO4(分析纯,江苏强盛公司); 多层硅胶商品柱、活性炭硅胶(日本Wako公司)。
PCDD/Fs标准样品:同位素稀释剂又称定量标,即含15个13C标记的2,3,7,8PCDD/Fs的混合标准溶液(Labeledcompound spiking solution,LCS),浓度为100/200 ng/mL; 进样内标溶液(Internal standards,ISS),即含浓度为200 ng/mL的13C1,2,3,4TCDD和13C1,2,3,7,8,9HxCDD的标准溶液,CS1~CS5校正标准溶液; PAR STOCK标准溶液; 仪器质量标准样品(Perfluorokerosene,PFK)。以上试剂均购于Wellington Laboratories 公司。
2.2 实验方法
2.2.1 样品预处理 将经过板器分离和多级破碎分选后所得的废线路板样品过60目筛,使废线路板和附着的元器件充分分离[17]。准确称取6.0 g过筛粉末于烧杯中,加入60 mL 2 mol/L HCl 溶液,去除样品中的炭状物等杂质,静置1 h后进行抽滤,以超纯水洗涤至中性后,用丙酮脱水。滤液用适量二氯甲烷滤液(1∶10, V/V)进行液液萃取3次,每次30 min,经无水Na2SO4脱水。
2.2.2 样品的提取 ASE法 将预处理后的滤渣、滤纸填入萃取池,加入适量硅藻土,添加5 μL提取内标溶液,萃取温度190℃,萃取压力1500 psi,甲苯60 mL,萃取池体积34 mL,循环次数3次,加热时间9 min, 静态萃取时间10 min。SE法:用新滤纸包裹预处理后的滤渣与滤纸,置于索氏提取管内,加入5 μL 提取内标溶液,用300 mL甲苯提取16 h以上。
2.2.3 样品的净化 依次采用多层硅胶柱和活性炭柱对萃取提取物进行净化[18,19]。多层硅胶商品柱的基本构成为石英棉/无水Na2SO4/硅胶/2% KOH硅胶/硅胶/44%硫酸硅胶/22%硫酸硅胶/硅胶/10%硝酸银硅胶/无水Na2SO4。用100 mL正己烷活化多层硅胶商品柱。将上述提取液与干燥后的萃取液合并,旋蒸浓缩至1~2 mL,转移至活化好的多层硅胶商品柱,用200 mL正己烷洗涤净化。活性炭柱能够选择性保留共平面结构的物质,如PCDD/Fs和PBDD/Fs等[4],其基本构成为石英棉/4 g无水Na2SO4/0.75 g活性炭/4 g无水Na2SO4。用20 mL正己烷活化活性炭柱。将多层硅胶商品柱净化后的淋洗液旋蒸至1~2 mL后,转移至活性碳柱净化。用200 mL 25%二氯甲烷正己烷混合溶液淋洗活性碳柱,再用125 mL甲苯淋洗,洗脱得到二噁英。
2.2.4 待测样品的制备 将甲苯洗脱液旋蒸至1~2 mL,转移至KD浓缩管中,氮吹至100 μL以下,用壬烷转移至带内衬管的进样小瓶中,添加5 μL进样内标溶液,混匀待测。
2.2.5 仪器分析 HRGCHRMS参数设置参考国标HJ 77.32008[20]及美国标准EPA 1613[11]。气相色谱参数:进样口温度为250℃,进样方式为不分流,进样量为1 μL,载气流量为1 mL/min。升温程序为:150℃保持3 min,20℃/min升至230℃,保持18 min,5℃升至250℃,保持10 min,4℃/min升至320℃,保持5 min。质谱条件为:PFK为质量参考物,电离方式为电子轰击离子化(Electron impact ionization,EI); 离子源温度为250℃; 加速电压为4.8 kV; 多离子检测(Multiple ion detector,MID)模式; 调谐仪器分辨率大于10000。
2.3 仪器检出限与方法检出限
选择制作相对响应因子的CS1~CS5 5个不同质量浓度点中最低质量浓度CS1进样1 μL,重复测定5次,测定溶液中2,3,7,8PCDD/Fs,检出限分别为四氯代五氯代二噁英类0.03~0.10 pg,六氯代七氯代二噁英类0.07~0.20 pg,八氯代二噁英类0.2~0.3 pg,满足EPA 1613[11]要求。取空白样品测定值3倍标准偏差计算方法检出限,ASE和SE对2,3,7,8TCDD的检出限分别为0.01和0.05 pg/g。
3 结果与讨论
3.1 ASE和SE的空白干扰
所用玻璃器皿均进行超声清洗,并使用甲醇、甲苯、二氯甲烷等依次润洗。ASE和SE操作和仪器空白干扰实验提取物为ASE萃取池填充物硅藻土,平行3次。17种2,3,7,8PCDD/Fs的定量分析结果表明,ASE和SE提取空白实验均未检出PCDD/Fs,说明操作过程及仪器中没有引入污染,实验流程对方法测定结果没有明显影响。
3.2 ASE和SE对空白样品中二噁英回收率的影响
分别运用ASE和SE进行方法空白实验,提取物为添加5 μL 15种提取内标物ASE萃取池填充物硅藻土,每种方法进行3次平行实验。空白样品中两种不同提取方法回收率及相对标准偏差,如图1所示。
分析结果表明,SE提取所得的加标回收率为65.7%~104.3%,相对标准偏差(Relative standard deviations,RSD)为3.0%~10.9%; ASE所得的加标回收率为58.6%~101.0%,RSD为1.2%~6.5%,均满足国标[20]要求。由图1可见,SE提取的平均加标回收率高于ASE,标准偏差低于ASE。对于空白样品,SE因样品基质简单、萃取步骤简便,且萃取时间长,准确度稍高。而ASE全过程均是仪器自动化运行,样品萃取过程较为稳定,对于基质单一的样品,ASE方法精密度更高[15,16]。此外,可以观察到两种提取方法对五氯代、六氯代二噁英/呋喃具有较高的回收率,四氯代、七氯代二噁英/呋喃次之,八氯代二噁英/呋喃的回收率最低[1,13]。值得注意的是,兩种提取方法对不同氯代数的多氯代二苯并二噁英和呋喃呈现显著不同的影响。多氯代二苯并二噁英氯代数的不同对其回收率的影响呈现峰状,而氯代数的不同对多氯代二苯并呋喃回收率的影响接近平稳的斜线。多氯代二苯并二噁英氯代数的不同对其回收率的影响明显大于多氯代二苯并呋喃。这一现象值得深入研究。
3.3 ASE和SE对实际样品中二噁英含量测定的影响
分别采取前述ASE和SE萃取实验步骤,对实际的废线路板样品中的二噁英和呋喃进行测定。实际样品中两种不同提取方法回收率及相对标准偏差如图2所示,SE提取所得的15种13C2,3,7,8PCDD/Fs的回收率范围为28.3%~77.7%,ASE所得的回收率范围为54.3%~113.0%,均满足国标[20]回收率要求(17%~181%)。ASE的回收率远高于SE,说明针对废线路板基质特点,ASE因其在高温高压下对样品的高渗透性和高萃取效率而具有较高的准确度,适用于废线路板中二噁英类物质的提取。另一方面,ASE的平均相对偏差为17.5%,高于SE平均相对偏差4.8%,表明SE在实际样品分析过程中仍明显具有良好重现性的优势,而ASE因样品的复杂性和萃取过程的简便快速性损失了一定的精密度。但在满足国际检测标准的范围内,ASE能够提供更为精确的污染物的含量,更适用于二噁英类物质的提取及含量的准确测定,尤其是对于废线路板等质地较为致密的样品,更能充分发挥ASE的高温高压的萃取条件优势。
由图2可知,两种不同的提取方法对15种13C2,3,7,8PCDD/Fs回收率的影响相似,多氯代二苯并二噁英和呋喃氯代数的不同对其回收率的影响分别呈现明显的峰状和轻微波动的斜线。其中五氯代、六氯代二噁英/呋喃回收率相对较高,八氯代二噁英/呋喃的回收率最低。多氯代二苯并二噁英氯代数的不同对其回收率的影响同样明显大于多氯代二苯并呋喃。
3.4 废线路板中二噁英含量的测定
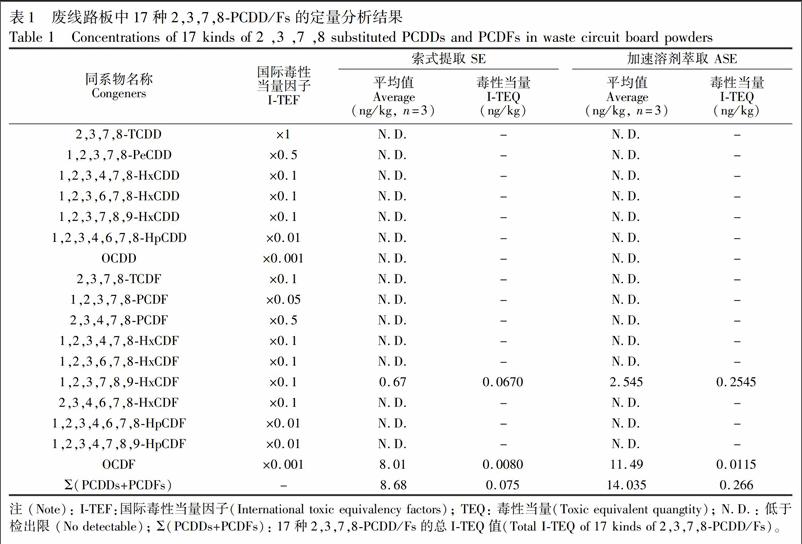
将废线路板样品分别采用SE和ASE萃取,再进行多层硅胶商品柱净化、旋蒸、氮吹浓缩后进行HRGCHRMS分析。二噁英的含量采用毒性当量(Toxic equivalent quangtity,TEQ)表示。TEQ质量浓度为实测质量浓度与该异构体的毒性当量因子的乘积。毒性当量因子(Toxic equivalent factors,TEF)指各二噁英类同类物与2,3,7,8TCDD对Ah受体的亲和性能之比[20]。由于选择的TEF体系不同,其计算的TEQ也不同[21]。分别测得样品中二噁英类总量为(TEQ) 0.0750 ng/kg和0.266 ng/kg,如表1所示。ASE所测得废线路板中二噁英毒性当量大于SE,说明在方法精密度允许范围内,ASE具有更好的萃取效果。
实测废线路板样品中1,2,3,7,8,9HxCDF浓度最高为0.067 ng/kg(TEQ)和0.2545 ng/kg(TEQ); OCDF为0.0080 ng/kg(TEQ)和0.0115 ng/kg(TEQ),其它均未检出。根据危险废物鉴别标准/毒性物质含量鉴别(GB 5085.62007)[22],实测废线路板中二噁英的含量远低于限值(15 μg/kg, TEQ),说明该批次废线路板的报废符合二噁英类物质的排放标准。
4 结 论
SE和ASE所得的废线路板中15种13C2,3,7,8PCDD/Fs的回收率均满足国标[20]要求,ASE对于废线路板中二噁英的萃取能力高于SE。ASE因在实际样品中能够发挥其高温高压的优势,加强了提取溶剂的渗透性进而提高了二噁英的萃取效率,较SE具有更高的准确度和合适的精密度。ASE代替SE可节省50%的溶剂,较大地缩短分析时间,更适用于大批量废线路板类复杂样品中超痕量二噁英类物质的提取。此外,因提取方法对不同氯代数的二噁英的萃取能力的不同,在处理含有不同氯代数的二噁英,尤其是含多氯代二苯并二噁英的样品时应考虑氯代数不同对提取效果的影响。
References
1 LI HuiRu, YU LiPing, ZHANG SuKun, REN Man, SHENG GuoYing, FU JiaMo, PENG PingAn. Chinese J. Anal. Chem., 2008, 36(2): 150-156
李会茹, 余莉萍, 张素坤, 任 曼, 盛国英, 傅家谟, 彭平安. 分析化学, 2008, 36(2): 150-156
2 Rimayi C, Chimuka L, Odusanya D, Boer J D, Weiss J. Chemosphere, 2015, 145: 314-21
3 Duan H B, Li J H, Liu Y C, Norimasa Y, Jiang W. Environ. Sci. Technol., 2011, 45(15): 6322-6328
4 XU PengJun, TAO Bu, LI Nan, ZHENG Sen, ZHAO Hu, FAN Shuang, ZHOU ZhiGuang, REN Yue, QI Li, CHEN JiPing. Chinese J. Anal. Chem., 2015, 43(3): 356-365
许鹏军, 陶 晡, 李 楠, 郑 森, 赵 虎, 范 爽, 周志广, 任 玥, 齐 丽, 陈吉平. 分析化学, 2015, 43(3): 356-365
5 Guéguen F, Stille P, Millet M. Environ. Sci. Pollut. Res. Int., 2013, 20(6): 3852-3862
6 DiasFerreira C, Kirkelund G M, Jensen P E. Chemosphere, 2016, 148: 380-387
7 Shin E S, Kim J, Choi S D, Kang Y W, Chang Y S. Chemosphere, 2016, 146: 419-425
8 Planche C, Ratel J, Mercier F, Blineta P, Debrauwerb L, Engel E. J. Chromatogr. A, 2015, 1392: 74-81
9 LI Xiang, ZHANG Yao, SUN YiZhi, ZHONG WeiKe, QIU YueMing, CHEN YanChang, WANG DaNing. Chinese J. Chromatogr., 2006, 24(4): 347-350
李 翔, 張 垚, 孙毅之, 仲维科, 邱月明, 陈彦长, 王大宁. 色谱, 2006, 24(4): 347-350
10 MA FeiPan, ZHANG SuKun, ZHAO BaoWei, REN MingZhong, XU Jie, QING Xian, ZHANG ManWen. Environmental Chemistry, 2014, 33(2): 243-247
马飞攀, 张素坤, 赵保卫, 任明忠, 徐 洁, 青 宪, 张漫雯. 环境化学, 2014, 33(2): 243-247
11 Method 1613B. Tetrathrough OctaChlorinated Dioxins and Furans by Isotope Dilution HRGC/HRMS. US EPA1997
12 ZHAO ShiYan, CUI ZhaoJie. Environ. Protec. Chem. Ind., 2006, 26(6): 518-521
赵士燕, 崔兆杰. 化工环保, 2006, 26(6): 518-521
13 ZHANG Yun, SU Jin, JIN ChengLong. Shanghai Journal of Preventive Medicine, 2006, 18(8): 368-372
張 昀, 苏 瑾, 金成龙. 上海预防医学杂志, 2006, 18(8): 368-372
14 Nie Z Q, Die Q Q, Yang Y F, Tang Z W, Wang G Q, Huang Q F. Environ. Sci. Pollut. Res., 2014, 21(13): 7863-7875
15 Berit B, Uta R. K, Norbert T. Environ. Sci. Pollut. Res., 2015, 22(24): 19887-19895
16 Zhang P, Ge L K, Zhou C G, Yao Z W. Chin. J. Oceanol. Limnol., 2011, 29(5): 1103-1112
17 MA JunWei, WANG ZhenZhen, YANG ZhiFeng, NIE YongFeng,BAI QingZhong. Techniques and Equipment for Environmental Pollution Control., 2005, 6(7): 63-66
马俊伟, 王真真, 杨志峰, 聂永丰, 白庆中. 环境污染治理技术与设备, 2005, 6(7): 63-66
18 GAO Dan, LIU JinSong, ZHU GuoHua, GONG HongPing, ZHOU Xin,WANG Ling, MO WeiMin, LI MuFei. Chinese J. Anal. Chem., 2013, 41(12): 1862-1868
高 丹, 刘劲松, 朱国华, 巩宏平, 周 欣, 王 玲, 莫卫民, 李霂菲. 分析化学, 2013, 41(12): 1862-1868
19 YU BinBin, FANG Cheng, RAO QinQuan, CHEN Tao, MOU YiJun, WU ZuCheng. Chinese J. Anal. Chem., 2011, 39(6): 833-838
余彬彬, 方 铖, 饶钦全, 陈 涛, 牟义军, 吴祖成. 分析化学, 2011, 39(6): 833-838
20 HJ 77.32008, Solid Waste Determination of Polychlorinated DibenzopDioxins(PCDDs)and Polychlorinated Dibenzofurans(PCDFs)Isotope Dilution HRGCHRMS. National Standards of the People's Republic of China
固体废物 二噁英类的测定 同位素稀释高分辨气相色谱高分辨质谱法. 中华人民共和国国家标准.HJ 77.32008
21 DU Bing, LIU AiMin, HUANG YeRu. Chinese Journal of Chromatography, 2004, 32(9): 867-970
杜 兵, 刘爱民, 黄业茹. 色谱, 2004, 32(9): 867-970
22 GB 5085.62007, Identification Standards for Hazardous WasteIdentification for Extraction Toxicity. National Environmental Protection Standard of the People's Republic of China
危险废物鉴别标准/毒性物质含量鉴别. 中华人民共和国国家环境保护标准. GB 5085.62007
