
分层结构微孔球扁药制备条件对其燃烧性能的影响
2017-05-08 01:06:22张一鸣丁亚军罗元香应三九
含能材料 2017年3期
张一鸣, 丁亚军, 罗元香, 应三九
(南京理工大学化工学院, 江苏 南京 210094)
1 引 言
小口径武器普遍采用钝感技术改善球扁药的减面性燃烧,即借助溶剂将阻燃剂渗入到发射药表层,降低该区域发射药的火焰温度和燃速,由于这些物质沿表面向内部方向浓度由大到小,燃速由小到大,最终变现为燃速的渐增性[1]。针对钝感技术普遍存在钝感剂选取工作量大、易迁移和燃烧产生大量烟焰等缺点,应三九课题组[2]提出利用超临界CO2技术制备分层结构微孔球扁药,可有效改善球扁药的燃烧性能。分层结构微孔球扁药内部微孔密度由外向内梯度递增,基于增燃面原理,在不引入钝感剂的前提下实现燃烧渐增性。
采用超临界CO2制备聚合物微发泡材料是材料领域的热点之一[3]。其制备过程的工艺参数,如饱和温度、饱和压力、饱和时间、发泡温度、发泡时间、卸压速率等对材料泡孔形态有着重要影响[4]。Reverchon[5]研究了聚苯乙烯(PS)的发泡过程,发现随着饱和压力的增大,泡孔密度增加,泡孔直径减小,泡孔尺寸分布变窄。Krause等[6]采用超临界CO2间歇升温法发泡聚醚酰亚胺(PEI)和聚醚砜树脂(PES),研究了发泡过程中的温度-浓度曲线,提出了发泡温度区间,并指出体系中CO2含量在一定的临界值以内才能形成闭孔结构。McCarthy课题组[7-8]针对卸压速率与泡孔结构之间关系作了深入研究,发现在很快的卸压速率下,泡孔直径和泡孔密度随卸压速率的增加而减小。
分层结构微孔球扁药的内外成分一致,因此其燃烧性能完全取决于内部结构。本研究在文献[2]的基础上,通过分析分层层微孔结构的形成机理,结合实验扫描电镜和密闭爆发器实验结果,研究了发泡工艺参数(解吸附时间和发泡温度)、内部结构和燃烧性能之间的对应关系。
2 实验部分
2.1 试剂与仪器
双基球扁药,255厂; CO2气体,纯度≥99.9%,南京文达特种气体有限公司; 液氮,文达特种气体有限公司; HA型超临界发泡装置,南通市华安超临界设备有限公司; QUANTA FEG250型扫描电镜,美国FEI公司; 50 mL密闭爆发器。
2.2 样品制备
采用快速升温发泡工艺[2],双基球扁药经过恒温恒压饱和、卸压解吸附、升温发泡和冷却定型,制得分层结构微孔球扁药样品。所有样品在恒温恒压阶段工艺参数相同: 压力ps=15 MPa、温度Ts=40 ℃。令球扁药达到吸收饱和后,卸压进入发泡阶段,发泡过程工艺参数如表1所示,0#样品为双基球扁药原药。
表1 发泡过程工艺参数
Table 1 Technological parameters of foaming process
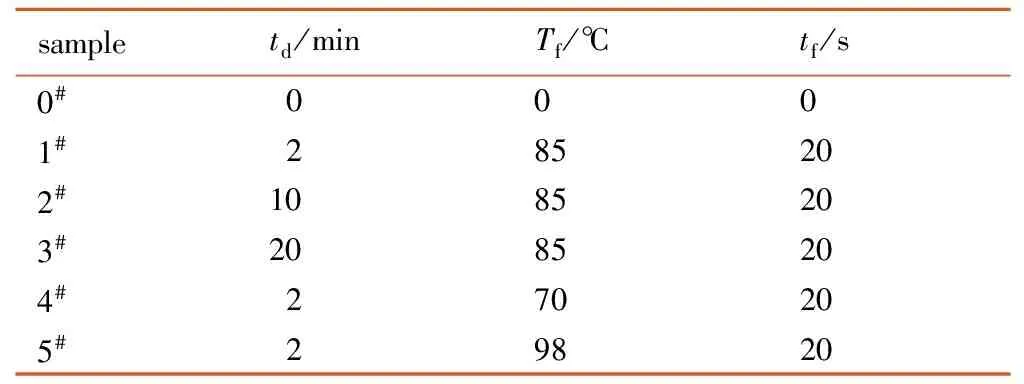
sampletd/minTf/℃tf/s0#0001#285202#1085203#2085204#270205#29820
Note:tdis desorption time,Tfis foaming temperature,tfis foaming time.
2.3 内部形貌表征
梯度微孔球扁药样品在液氮中浸泡10min后脆断,表面喷金,利用扫描电镜对其断面进行观察。
2.4 燃烧性能表征
利用密闭爆发器实验测试样品和双基球扁药的燃烧性能。实验测试温度为25 ℃,密闭爆发器容积为50 mL,测试样品装填密度为0.12 g·cm-3,点火压力为9.8 MPa,点火药为2#硝化棉。每种测试样品进行三次实验,得到p-t曲线,并对其进行处理得到L-B曲线。
3 结果与讨论
3.1 皮层形成机理
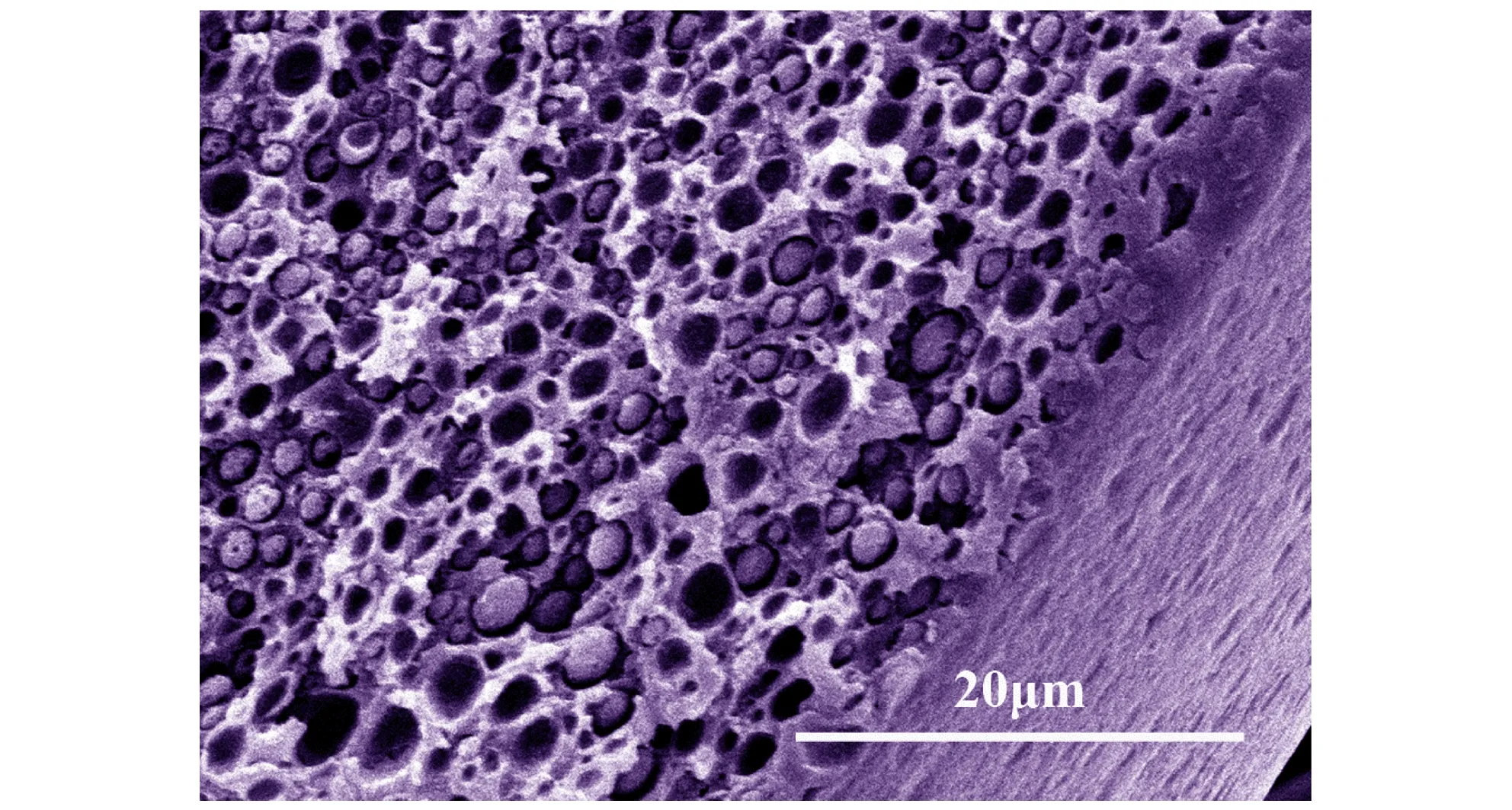
对发泡前后的样品进行断面边缘SEM表征,结果如图1所示,由图1可见,未发泡样品内部整体为密实结构(图1a),经过发泡处理的样品(图1b)外侧为皮层,该区域样品未发泡(或泡孔密度很低); 样品内部为发泡层,该区域存在大量致密泡孔。这说明可以通过调节发泡过程的工艺参数制备内部具有分层结构的微孔球扁药。
假设球扁药样品表层平整,且CO2吸附扩散系数恒定,距离样品中心x处在解吸附时间td后的气体浓度可用式(1)进行描述[10]:
(1)
式中,C为距离样品中心x处,解吸附时间td时的气体浓度,mol·L-1;C0为解吸附开始前的气体起始浓度,mol·L-1;C1为样品表层气体浓度,mol·L-1;D为扩散系数;td为解吸附时间,min;L为样品厚度,cm;x为距离样品中心的距离,cm。

a. unfoamed
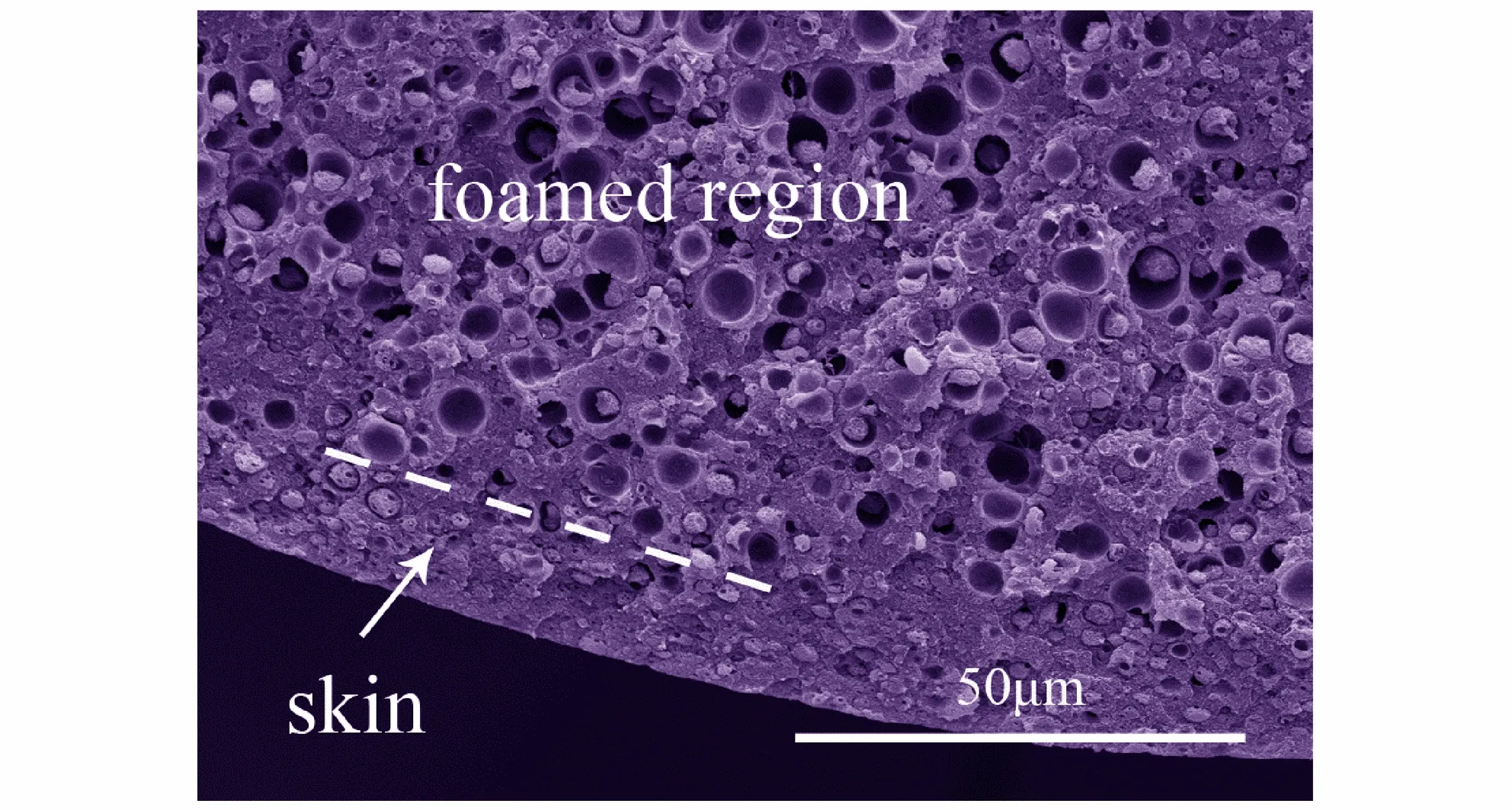
b. foamed
图1 原药和分层结构微孔球扁药的断面SEM图
Fig.1 SEM images of fracture surface of original propellant and microcellular oblate spherical propellants with layered structure
样品置于自然环境下时,表层气体浓度可以忽略不计。令C1=0,则式(1)可以简化为:
(2)
未发泡皮层的形成机理如图2所示。在一定的发泡温度Tf下,样品存在一个最低发泡浓度C*,只有CO2浓度大于C*的区域才有可能形成大量致密的泡
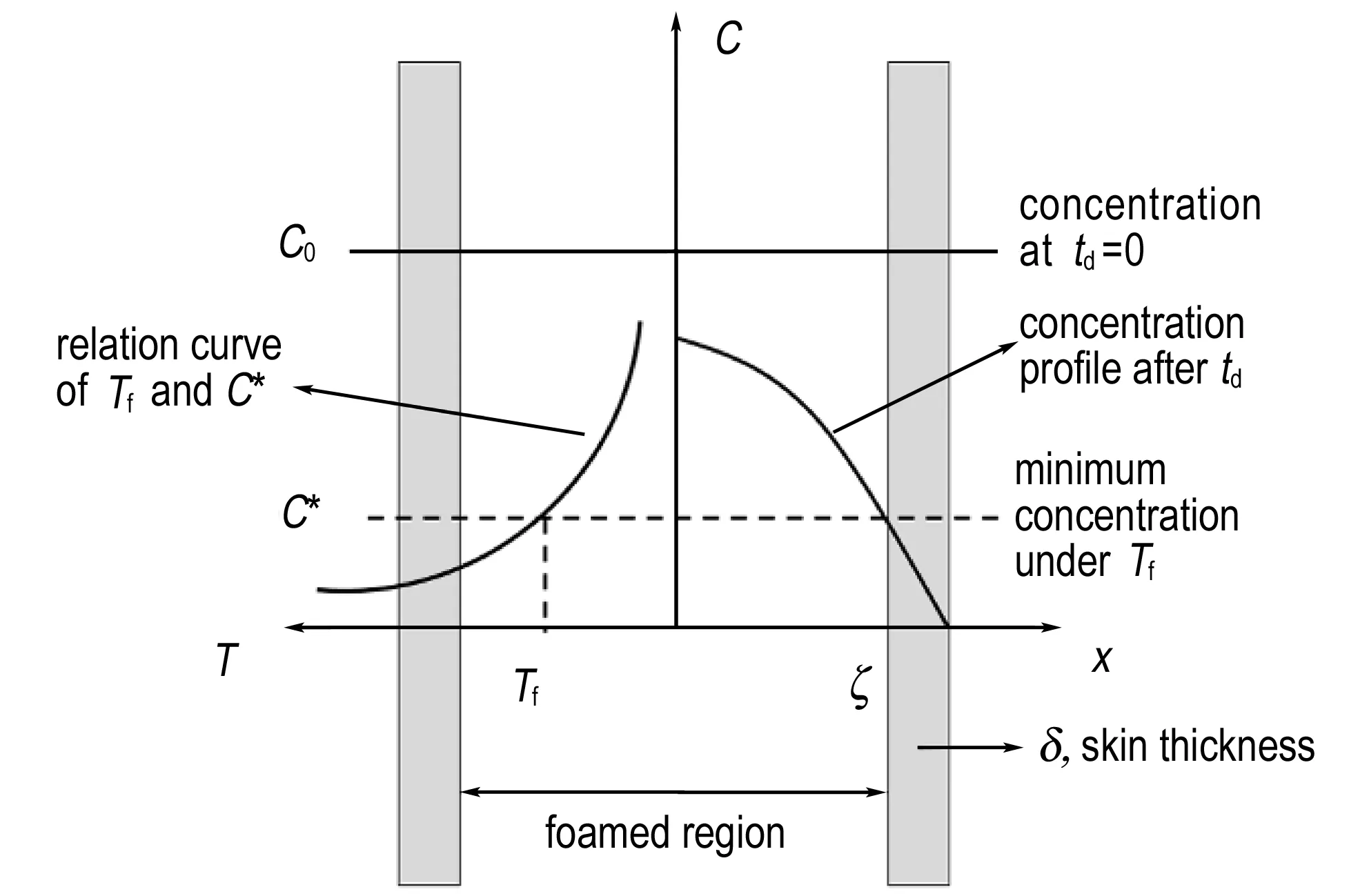
图2 未发泡皮层形成机理
Fig.2 Forming mechanism of the unfoamed skin
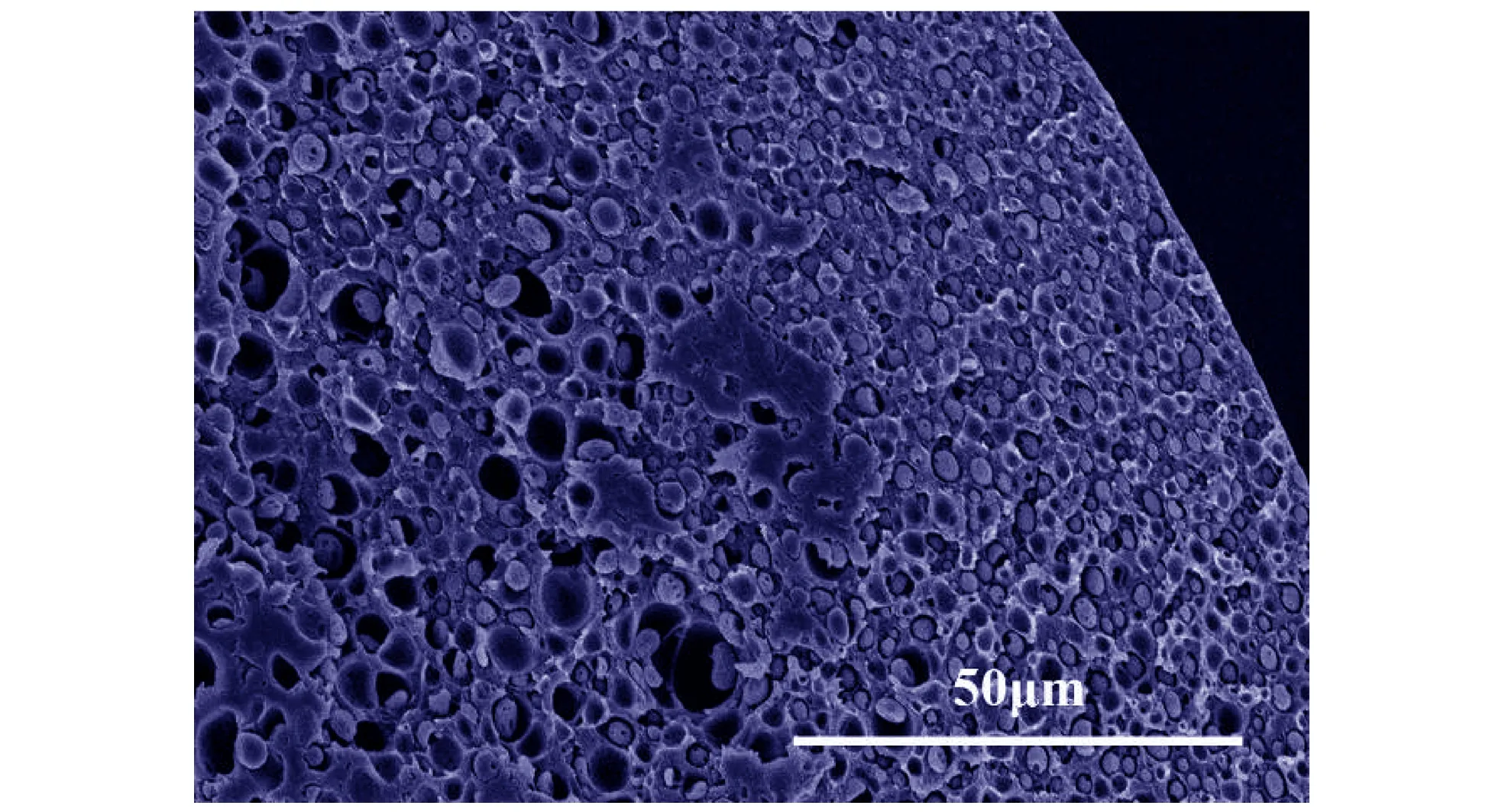
孔。利用升温工艺时,卸压后的解吸附过程会造成CO2从已达到吸收平衡的球扁药/CO2体系中逃逸。根据式(2),解吸附td时间后样品中CO2浓度由内至外呈梯度递减分布。当td时刻CO2浓度分布曲线与直线C=C*相较于x=ζ时,厚度δ的皮层就形成了。样品内层(x<ζ)CO2浓度高于C*,发泡形成致密泡孔,是发泡层; 样品外层(ζ δ=L/2-ζ (3) 不同解吸附时间下制备的样品(1#,2#,3#)内部形貌如图3所示。从图3a可以看出,当解吸附时间为2 min时,1#样品皮层厚度约为20 μm,而内部则有大量致密泡孔且分布均匀。由图3b可知,2#样品的解吸附时间为10 min,其皮层厚度约为50 μm,内部泡孔密度与1#样品相比明显降低。这是因为解吸附时间的增加导致样品内部CO2整体浓度降低,泡孔生长动力下降。由图3c可见,解吸附时间为20 min时,3#样品皮层在不同区域的厚度有所差异(可能的原因是原双基球扁药组分分布不均,从而导致各区域在饱和过程中溶解的CO2含量不同),但整体皮层厚度仍大于2#样品。对比图3a、图3b、图3c,发现在其他条件相同时,随着解吸附时间td的增加,样品皮层厚度增大。由式(2)和式(3)可知,当起始浓度C0一定时,若C=C*不变,随着解吸附时间td增加,ζ值减小,即更加靠近样品中心的区域才能达到最低发泡浓度,因此皮层δ厚度增加。 a. 1#(2 min) b. 2#(10 min) c. 3#(20 min) 图3 不同解吸附时间样品的断面SEM图 Fig.3 SEM images of fracture surface of samples with different desorption times 解吸附2 min后在不同发泡温度下制备的样品(4#,5#)内部形貌的SEM结果如图4所示。由图4可见,当发泡温度为70 ℃时,4#样品(图4a)仅有中心区域发泡明显,其他区域几乎没有泡孔存在,皮层厚度大于100 μm。而从图4b可以看出,当发泡温度为98 ℃时,5#样品边缘区域发泡明显,没有形成有效的皮层。由图4a,图4b与图3a对比可知,在解吸附时间相同时(td=2 min),随着发泡温度的增加,皮层厚度逐渐减小,最终未能形成皮层结构。 a. 4# (70 ℃) b. 5# (98 ℃) 图4 不同发泡温度样品的断面SEM图 Fig.4 SEM images of fracture surface of samples with different foaming temperatures 泡孔成核是微孔结构形成的关键步骤。分层结构微孔球扁药样品中系中的气体分子因热力学不稳定(升高温度)聚集成为临界气核,当气体分子继续向气核扩散时,泡孔开始形成。体系的成核速率N(单位时间、单位体积内生成气核的数目)可表示为[10-11]: N=C·f·exp(-ΔG/kTf) (4) 式中,C为成核点的CO2气体浓度,mol·L-1;f为频率因子; ΔG为成核时所需克服的活化能垒;k为Boltzmann常数;Tf为发泡时体系的绝对温度,K。 可以认为当区域内成核速率N达到某一定值Nf时,该区域才能形成具有大量致密泡孔的发泡层。在一定的发泡温度Tf下,成核速率达到Nf时的成核点浓度即为最低发泡浓度C*。由式(4)可知,若N=Nf,当Tf升高时,C*降低,而结合图2发现,较低的C*会导致皮层厚度δ减小,与扫描电镜结果相吻合。 此外,泡孔成核后气泡的生长需满足如下关系: Tf≥Tlower=Tg (5) 式中,Tf为发泡温度;Tlower为聚合物/气体混合体系最低发泡温度;Tg为聚合物/气体混合体系玻璃化温度。 可见,只有当发泡温度大于体系的玻璃化温度时,才能形成泡孔结构。聚合物中溶解的CO2会降低聚合物/气体混合体系的玻璃化温度,在一定范围内可用式(6)描述[12]: (6) 因此,4#样品(图4a)中很大区域未发泡可能是以下两个原因共同作用的结果: 一是样品中CO2浓度低于Tf=70 ℃时的C*,无法形成足够数量的气核; 二是未发泡皮层区域溶解的CO2没有使Tg低于70 ℃,泡孔不能生长。 采用密闭爆发器实验研究分层结构微孔球扁药的燃烧性能。根据u-t曲线数据计算得动态燃烧活度L和相对压力B。双基球扁药原药(0#)和不同解吸附时间样品(1#,2#,3#)的L-B曲线如图5所示。由图5可见,所有样品L值均有不同程度的增大,解吸附时间td越小,L值增大越明显。B<0.2时,各样品L值与原药相比差距较小,且均呈递增趋势。在0.2 图5 不同解吸附时间样品和原药的L-B曲线 Fig.5L-Bcurves of original propellant and samples with different desorption times 双基球扁药原药(0#)和不同发泡温度样品(1#,4#,5#)的L-B曲线如图6所示。4#样品(Tf=70 ℃)与原药基本相同,没有改善发射药的燃烧性能。5#样品(Tf=98 ℃)的L值明显高于其他样品,说明该样品燃烧最为剧烈。L值在起始阶段迅速增大,增大幅度远高于原药和其他样品,并在B=0.25时达到峰值。但是,L值在B>0.25后迅速降低,样品的燃烧渐增性改善并不明显。 图6 不同发泡温度样品和原药的L-B曲线 Fig.6L-Bcurves of original propellant and samples with different foaming temperatures 结合样品内部形貌(图2,图3)发现,样品的皮层厚度很大程度上决定了其燃烧渐增性。样品1#表现出良好的燃烧渐增性,说明此厚度(约20 μm)的皮层较为合理。2#样品、3#样品、4#样品的皮层厚度逐渐增大,燃烧渐增性改善效果也越来越差。这是因为样品的燃烧可分为两个过程: 一是密实皮层,燃烧性能与原药同为减面燃烧,; 二是内部微孔发泡层,燃烧面积骤然增加,燃气释放速率增大,燃烧活度增加。皮层厚度越大,内部增面燃烧的发泡区域就越小,最终导致样品燃烧渐增效果降低。5#样品完全发泡,没有形成燃速较低的皮层,样品内外泡孔密度的梯度差异不明显,发射药燃气释放速率递增不足,因此也没有表现出良好的燃烧渐增性。 (1) 在发泡时间不变的条件下,皮层的形成受到解吸附时间和发泡温度的影响。 (2) 解吸附时间和发泡温度可以控制样品的皮层厚度。解吸附时间越长,皮层厚度越大; 发泡温度越高,皮层厚度越小,当发泡温度过高时(98 ℃),未形成皮层。 (3) 皮层厚度很大程度上决定了分层结构微孔球扁药的燃烧性能。对于一定尺寸的球扁药,某个或某个范围内的皮层厚度才能使其具有燃烧渐增性。在解吸附时间td=2 min、发泡温度Tf=85 ℃、发泡时间tf=20 s条件下制备的样品(皮层厚度约为20 μm)表现出了良好的燃烧渐增性。 参考文献: [1] 王泽山, 何卫东, 徐复铭. 火药装药设计原理与技术[M]. 北京: 北京理工大学出版社, 2006: 210-219. WANG Ze-shan, HE Wei-dong, XU Fu-ming. Design principle and technique of powder charge[M]. Beijing: Beijing Institute of Technology Press, 2006: 210-219. [2] 陈西如, 应三九, 肖正刚. 超临界CO2制备微孔球扁药的研究[J]. 兵工学报,2012, 33(5): 534-539. CHEN Xi-ru, YING San-jiu, XIAO Zheng-gang. Research on preparation of microfoam oblate spherical propellants by supercritical CO2[J].ActaArmamentarii, 2012, 33(5): 534-539. [3] Tomasko D L, Burley A, Feng L, et al. Development of CO2for polymer foam applications[J].TheJournalofSupercriticalFluids, 2009, 47(3): 493-499. [4] 翟文涛, 余坚, 何嘉松. 超临界流体制备微发泡聚合物材料的研究进展[J]. 高分子通报, 2009(3): 1-10. ZHAI Wen-tao, YU Jian, HE Jia-song. Research progresses in preparation of microcellular polymers by supercritical fluid technique[J].PolymerBulletin, 2009(3): 1-10. [5] Reverchon E, Cardea S. Production of controlled polymeric foams by supercritical CO2[J].TheJournalofSupercriticalFluids, 2007, 40(1): 144-152. [6] Krause B, Sijbesma H J P, Münüklü P, et al. Bicontinuous nanoporous polymers by carbon dioxide foaming[J].Macromolecules, 2001, 34(25): 8792-8801. [7] Arora K A, Lesser A J, McCarthy T J. Preparation and characterization of microcellular polystyrene foams processed in supercritical carbon dioxide[J].Macromolecules, 1998, 31(14): 4614-4620. [8] Arora K A, Lesser A J, McCarthy T J. Compressive behavior of microcellular polystyrene foams processed in supercritical carbon dioxide[J].PolymerEngineering&Science, 1998, 38(12): 2055-2062. [9] Kumar V, Weller J E. A model for the unfoamed skin on microcellular foams[J].PolymerEngineering&Science, 1994, 34(3): 169-173. [10] Colton J S, Suh N P. The nucleation of microcellular thermoplastic foam with additives: Part I: Theoretical considerations[J].PolymerEngineering&Science, 1987, 27(7): 485-492. [11] Colton J S, Suh N P. The nucleation of microcellular thermoplastic foam with additives: Part II: Experimental results and discussion[J].PolymerEngineering&Science, 1987, 27(7): 493-499. [12] Krause B, Mettinkhof R, Van der Vegt N F A, et al. Microcellular foaming of amorphous high-Tgpolymers using carbon dioxide[J].Macromolecules, 2001, 34(4): 874-884.3.2 解吸附时间对皮层厚度的影响


3.3 发泡温度对皮层厚度的影响

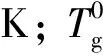
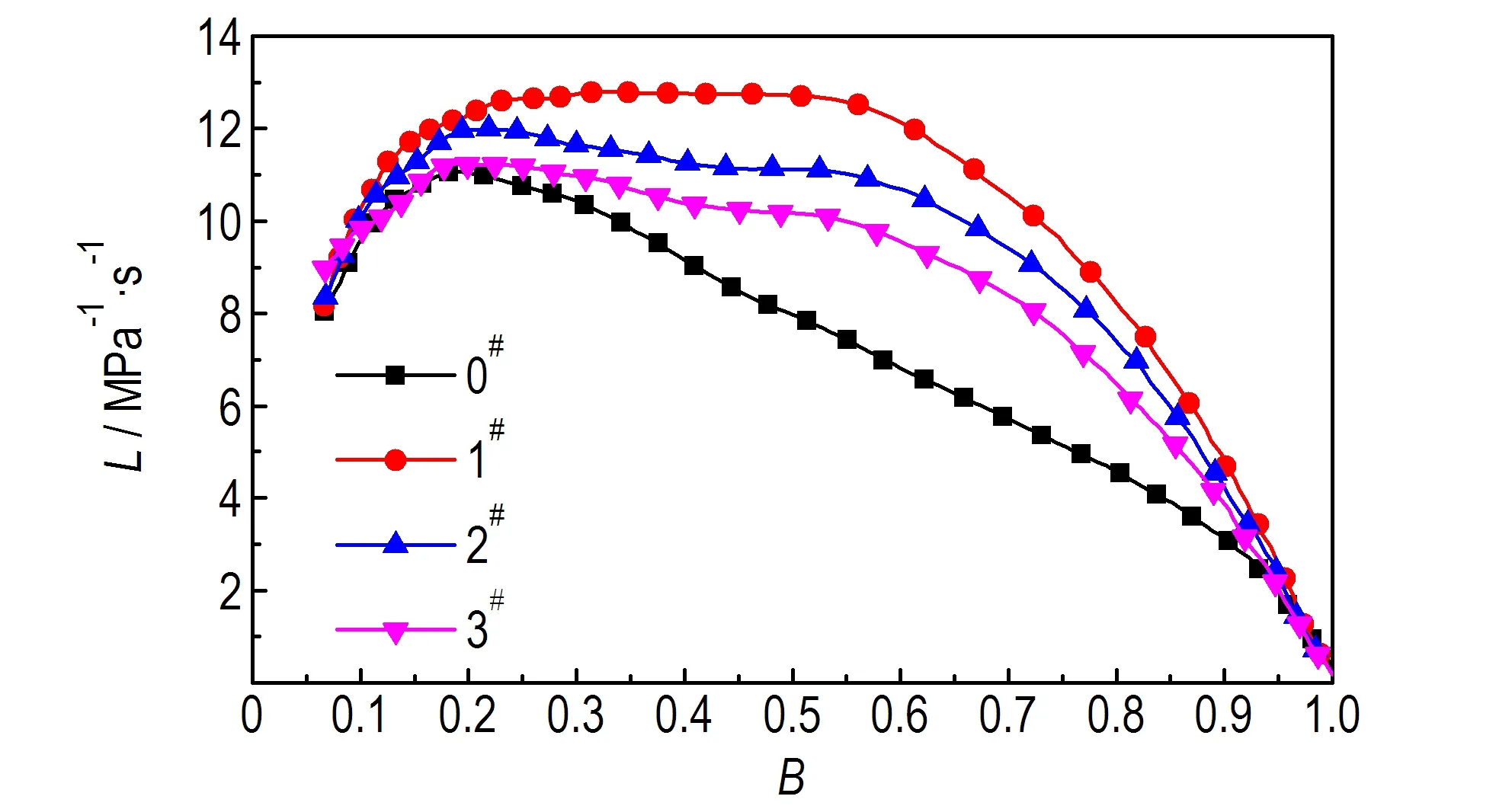
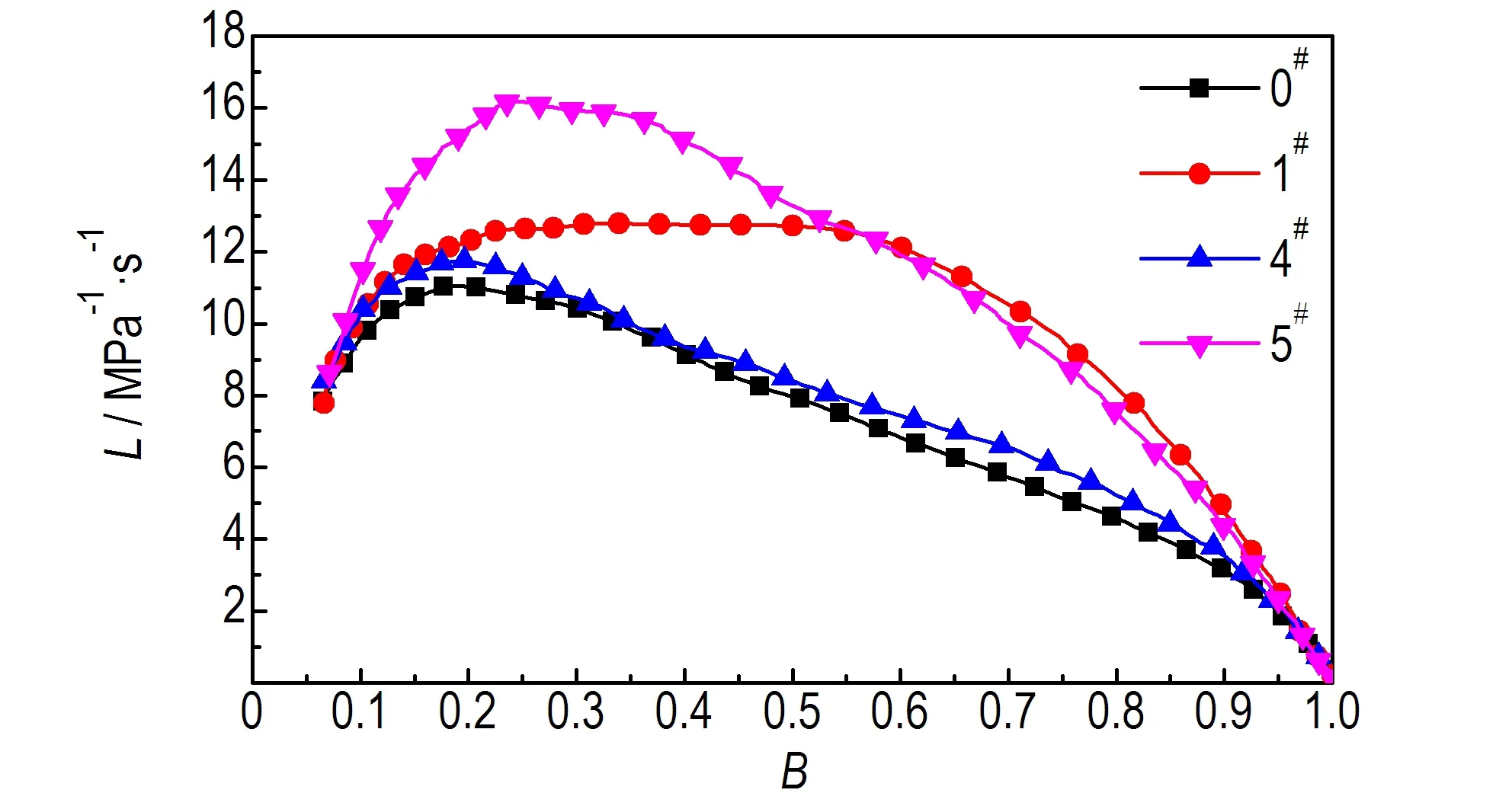
3.4 样品的燃烧性能测试
4 结 论
猜你喜欢
包装工程(2022年1期)2022-01-26 09:03:10科教导刊·电子版(2021年6期)2021-05-06 05:05:14工程塑料应用(2020年11期)2020-11-28 01:57:50中国农资(2016年31期)2016-09-13 00:59:13中国农资(2016年34期)2016-08-11 08:33:46中国农资(2016年36期)2016-08-09 01:06:10中国农资(2016年33期)2016-08-02 08:41:23中国塑料(2016年4期)2016-06-27 06:33:34中国塑料(2015年3期)2015-11-27 03:42:12中国塑料(2015年7期)2015-10-14 01:02:44
