
深度学习在化学信息学中的应用
2017-04-21 08:06:35徐优俊裴剑锋
大数据 2017年2期
徐优俊,裴剑锋
北京大学前沿交叉学科研究院定量生物学中心,北京 100871
深度学习在化学信息学中的应用
徐优俊,裴剑锋
北京大学前沿交叉学科研究院定量生物学中心,北京 100871
深度学习在计算机视觉、语音识别和自然语言处理三大领域中取得了巨大的成功,带动了人工智能的快速发展。将深度学习的关键技术应用于化学信息学,能够加快实现化学信息处理的人工智能化。化合物结构与性质的定量关系研究是化学信息学的主要任务之一,着重介绍各类深度学习框架(深层神经网络、卷积神经网络、循环或递归神经网络)应用于化合物定量构效关系模型的研究进展,并针对深度学习在化学信息学中的应用进行了展望。
深度学习;人工智能;定量构效关系;化学信息学
1 引言
人工智能(artificial intelligence,AI)主要用人工的方法在机器(计算机)上实现智能化,或称机器智能。自2006年以来,机器学习领域取得了突破性的进展。深度学习(deep learning)算法的提出让人们相信AI离人们并没有那么遥远。自AlphaGo战胜李世石的那一刻起,深度学习技术被认为是目前最有可能实现AI的技术之一。深度学习是机器学习算法的一种。在最近的10年中,深度学习技术在语音识别[1]、计算机视觉[2]和自然语言处理[3]中取得了巨大的成功。同时,该技术凭借其强大的能力也逐渐地被应用到化学信息学的很多领域[4],如计算机辅助药物设计、材料性质的预测等。在2012年的Merck公司组织的药靶化合物活性预测比赛中,利用深度学习技术构建的预测模型不仅战胜了所有的竞争者,而且在准确率方面大约超出Merck公司内部评价系统15%。随后,利用深度学习开发的多任务模型在2014年美国国立卫生研究院(National Institutes of Health,NIH)组织的Tox21毒性预测挑战赛中也获得了令人欣喜的成绩。在活性和毒性的预测方面的成功,让研究人员产生了疑问,深度学习算法比起传统机器学习算法,优势在哪?首先对于传统的机器学习算法来说,有3个特点:特征是人为设定的,在特征提取或者构建过程中,不同的转换和近似被应用到输入特征上;简单的模板匹配,只考虑如何利用输入特征集合产生较为满意的结果,并不注重对问题的表征;模型的表现力随着参数的增多而逐渐变强。而深度学习通过多层人工神经网络(artificial neural network,ANN)将输入特征进行不同层次水平的表征,实现“分层表征”的概念,进而通过多种训练技巧优化模型的表现。其中每一层的表层也有可能被应用到其他新的问题中。模型的表现力随着层数的增多而呈指数增长[5]。深度学习凭借这样的特性能够直接在输入原始数据的基础上进行特征训练,最后得出具有竞争力的预测模型。简而言之,深度学习算法不仅能够构建预测模型,而且具有潜在的自动学习特征的能力。
化学信息学(chemoinformatics)是一门应用信息学方法解决化学问题的学科。其主要任务之一是基于化合物二维(2D)或者三维(3D)结构发展能够预测化合物潜在性质的模型,该模型基于的基本假设是“相似的分子具有相似的性质”。这一假设也被称为构效关系(structureacitivity relationship,SAR),用来建立化合物结构与性质之间的相关性。定量构效关系(quantitative structure-acitivity relationship,QSAR)是在构效关系的基础上,结合物理化学中常用的经验方程的数学方法出现的,其理论历史可以追溯到1868年提出的Crum-Brown方程,该方程认为化合物的生理Φ可以用化学结构C的函数表示,但是并未建立明确的函数模型。最早可以实现的定量构效关系方法是由美国波蒙拿学院的Hansch在1964年提出的Hansch方程:。其中,lgP是疏水参数,ES是立体参数,σ是电性参数,a、b、ρ、d是方程系数。Hansch模型揭开了经典QSAR研究的篇章,成为QSAR发展历史中的里程碑。其后QSAR的研究被广泛用于预测化合物的药代动力学性质,如吸收、分布、代谢、排泄和毒性。构建QSAR模型一般需要3个主要步骤:生成已知化合物测量性质的训练集;编码关于化合物的化学结构的信息;建立数学模型,从编码的化学结构信息预测测量的性质。高通量筛选(high throughput screening,HTS)是收集训练数据的理想方法。针对感兴趣的分子性质,可以设计高通量的芯片来完成几百到数千种化合物测试,这些被测量的性质可以是细胞或生物化学的表型。目前已经有很多将化合物编码成数字向量的方法,这些方法主要针对分子的各种物化性质和拓扑性质进行描述。各种机器学习方法被用来构建数学模型,从而进行化合物预测。这些方法包括线性回归模型、贝叶斯神经网络、随机森林(random forest,RF)、偏最小二乘和支持向量机(support vector machine,SVM)等。在面对一些复杂的分子性质预测时,传统的机器学习算法仍有所不足,而深度学习技术凭借其突出的能力为化学信息学研究增添了一道新的曙光。
当前大多数的深度学习算法是基于人工神经网络发展而来的,在这里着重介绍具有深层网络架构的深度学习算法在QSAR研究中的应用进展情况。首先介绍深度学习的基础背景以及近些年的发展状况;然后介绍各类深度学习框架在QSAR中的应用实例,对目前存在的传统机器学习模型与深度学习模型的预测表现进行比较与分析;再将所有提到的方法进行汇总和比较分析;最后对深度学习在QSAR中的应用进行总结与展望,对化学信息的人工智能化进行展望。
2 深度学习
2.1 深度学习的背景和面临的障碍
深度学习的概念源于人工神经网络。人工神经网络是基于生物学中神经网络的基本原理,在理解和抽象了人脑结构和外界刺激响应机制后,以网络拓扑知识为理论基础,模拟人脑的神经系统对复杂信息的处理机制的一种数学模型。ANN是一种运算模型,由大量的节点(或称神经元)相互连接构成,如图1(a)所示。每个节点代表一种特定的输出函数,称为激活函数(activation function),每两个节点之间的连线代表权重(weight)。为了最小化预测误差的目标,ANN模型中的权值都将随着不断的训练而被调整。该网络由3部分构成:输入层(L1)、隐层(L2)和输出层(L3)。3维的输入信息通过各层神经元的非线性组合映射到1维的输出值。其中偏置项是指输入值为1的神经元。深层神经网络(deep neural network,DNN)架构示意如图1(b)所示,该网络架构包含多个隐层(如L2、L3)。
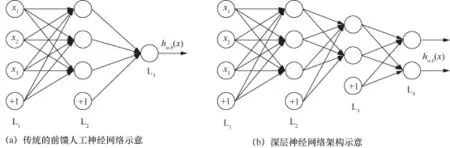
图1 人工神经网络架构示意
在前文提到,ANN模型的表现能力依赖于隐层的多层非线性转换能力[5]。随着层数的增多、层宽(每层神经元数目)的增大,更加复杂和抽象的特征将被组建,相应地,模型也能够学到更加复杂和抽象的表征。在面对多隐层的ANN如何调整参数的问题时,反向传播(backpropagation,BP)算法在ANN中发挥了极其关键的作用。BP算法根据输出的误差利用梯度下降算法对权值进行反向调整。尽管BP算法的概念在1963年就被提出,但是直到1986年,Hinton等人才将该算法应用到ANN模型的训练中,使得该方法成为了一套实用的工具。但是BP算法在反向传播时,梯度随着隐层数的增加越来越扩散,进而导致接近输入层的权值比较小,真正起到决策作用的仅仅是接近输出层的权值,导致模型的过拟合。这就是通常所说的“梯度弥散问题”。或许在2006年以前训练DNN最大的障碍就是梯度弥散问题,因为它几乎覆盖了人工神经网络的每一层。当然在模型比较复杂时,过拟合问题也是一个不可忽视的问题(如图2所示),在训练集上表现很好,但在测试集上就变得很差劲。图2中虚线表示的模型就一个过拟合的模型。
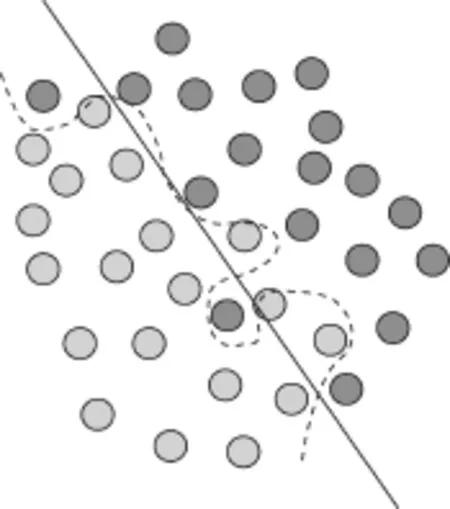
图2 过拟合问题示意
2.2 深度学习的训练技术
DNN模型面临的困境迫使研究人员发展新的训练算法,试图解决梯度弥散问题和降低过拟合问题。目前这些算法主要有:无监督预训练[6]、修正的线性函数(ReLU)、Dropout[7]、DropConnect和BatchNormalization[8]。
2006年由Hinton G E等人[6]开发的无监督预训练方法是一套快速贪婪的DNN训练算法。该算法先对DNN逐层进行无监督的预训练,然后利用随机梯度下降,微调整个网络。由于使用预训练之后,模型在BP之前已经学到了一些特征,然后再进行微调,能够很好地缓和梯度弥散的问题。随后Bengio Y等人[9]在2011年提出了ReLU激活函数,该函数被实验证明能够很好地规避梯度弥散问题。与Sigmoid函数(如图3(a)所示)不同,ReLU函数的一阶导数不是0就是1,如图3(b)所示,这样确保了误差能够较好地反向传递,梯度不会消失。在克服梯度弥散的同时,Dropout和DropConnect技术被发展用来降低过拟合的问题。Dropout是在训练过程中随机地屏蔽一定比例的神经元,使其不参与模型前馈的计算。该方法相当于在训练过程中不断改变模型的架构,防止神经元之间相互依赖关系,从而降低过拟合[7]。DropConnect与Dropout类似,是通过随机地屏蔽一定比例的权值,降低权值之间相互依赖关系,从而降低过拟合[7]。在兼顾梯度弥散和过拟合问题时,将前面提到的ReLU和Dropout技术同时应用到DNN的训练中,通常能够较好地矫正模型,从而提高模型的竞争力。BatchNormalization技术是谷歌公司在2015年提出的一套训练方法,在训练过程中加入了对小批量的数据集进行归一化的处理操作,从而提高模型的性能。其优势主要有以下几点。
● 允许使用更高的学习率提高训练速度。一般来说,较高的学习率能够引起梯度的弥散。在模型训练过程中,如果每层的尺度不一致,每层所需要的学习率是不一样的,同一层不同维度的尺度往往也需要不同大小的学习率,为此需要使用最小的学习率才能保证损失函数有效下降,BatchNormalization技术将每层、每维度的尺度保持一致,可以直接使用较高的学习率进行优化。
● 允许移除或使用较低的Dropout。Dropout是上述提到的常用的防止过拟合的方法。本文在测试中分别使用10%、5%和0的Dropout来训练模型,与之前40%~50%的Dropout相比,可以大大提高训练速度,并且维持模型原有的表现[8]。所以BatchNormalization技术是一套兼顾梯度和过拟合问题的加速训练方法。
以上是对当前深度学习训练技术的一些简介,当然还有很多其他的技术,比如Adagrad[10]、Adadelta[11]、RMSprop、Adam[12]等各类随机梯度下降技术。
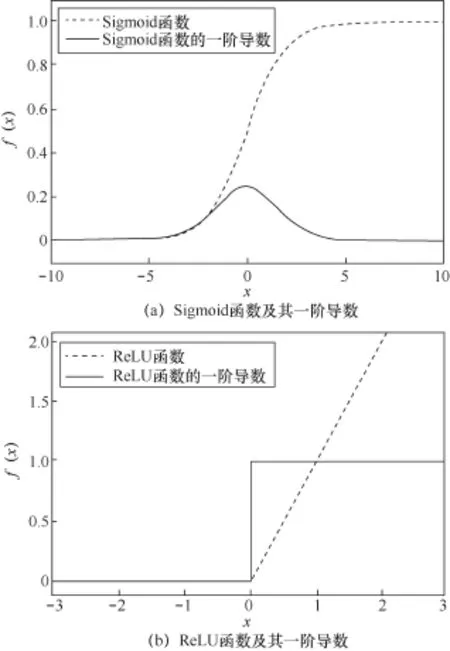
图3 Sigmoid函数和ReLU函数及其一阶导数
2.3 深度学习的模型架构
深度神经网络的架构有很多,其中最主要的除了前文提到的深层神经网络架构(如图1 (b)所示),还有一些应用在计算机视觉和自然语言处理中较为成功的深度网络架构,有卷积神经网络(convolutional neural network,CNN)(如图4 (a)所示)[2]、循环或递归神经网络(recu siveorrecur rentneural network,RNN)(如图4 (b)所示)[13,14]、自编码器(autoencoder,AE)(如图4 (c)所示)[15]。
图4 (a)中卷积操作有助于局部的空间信息的提取,从而学习到较好的表征;图4 (b)主要是将上一次迭代的输出作为当前迭代的输入,从而实现循环;图4 (c)主要通过逐层的无监督的学习将输入数据进行压缩表征,然后通过全连接的网络进行有监督的学习;图4(d)是将一张2 000多维的图像压缩成30维的表征过程。
CNN的基本结构包括两层,其一为特征提取层,每个神经元的输入与前一层的局部接收域相连,并提取该局部的特征。一旦该局部特征被提取后,它与其他特征间的位置关系也随之确定下来。其二是特征映射层,网络的每个计算层由多个特征映射组成,每个特征映射是一个平面,平面上所有神经元的权值相等。CNN主要用来识别位移、缩放及其他形式扭曲不变性的2D图形。CNN以其局部权值共享的特殊结构在语音识别和图像处理方面有着独特的优越性,其布局更接近于实际的生物神经网络,权值共享降低了网络的复杂性,特别是多维输入向量的图像可以直接输入网络这一特点,避免了特征提取和分类过程中数据重建的复杂度。
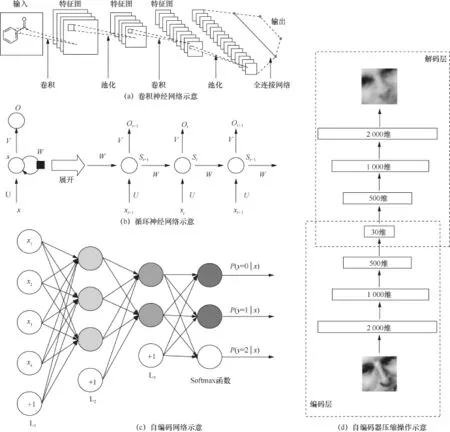
图4 其他深度神经网络架构
RNN适合处理序列数据。传统的神经网络模型是从输入层到隐层再到输出层,层与层之间是全连接的,每层之间的节点是无连接的。但是这种普通的神经网络对于很多问题却无能无力。例如,要预测句子的下一个单词是什么,一般需要用到前面的单词,因为一个句子中前后单词并不是独立的。RNN之所以称为循环神经网络,是因为一个序列当前的输出与前面的输出也有关。具体的表现形式为网络会对前面的信息进行记忆,并应用于当前输出的计算中,即隐层之间的节点不再是无连接的,而是有连接的,并且隐层的输入不仅包括输入层的输出,还包括上一时刻隐层的输出。RNN已经在实践中被证明其对自然语言处理是非常成功的,如词向量表达、语句合法性检查、词性标注等[16]。在RNN中,目前使用最广泛最成功的模型便是长短时记忆(long short-term memory,LSTM)[17]模型。
AE是一种无监督的机器学习技术,利用神经网络产生的低维数据代表高维输入(如图4(d)所示)。传统上,依靠线性降维方法(如主成分分析(principal component analysis,PCA))找到在高维原始数据上最大方差方向,通过选取部分较大方差的轴来实现降维。PCA的目的是捕获包含大部分信息输入的方向,用尽可能小的维度表征原始输入。然而,PCA的线性组合限制了复杂特征的提取,AE则用固有的非线性神经网络克服了这些限制。AE由两个主要部分组成:编码层和解码层。编码层在训练和部署时被使用,解码层只是在训练时使用。编码层的目的是找到一个给定维度的压缩表征,通过逐层的无监督预训练优化低维的表征。解码层是编码层的一个反射,用来重建尽可能密切的原始输入。利用这样的方法进行训练的目的是迫使AE选择最丰富的特征压缩路线,尽可能逼近原始输入。
以上是对深度神经网络3个主要框架的简介。还有很多变体的架构大多是基于这三者发展而来的。
2.4 深度学习的自动特征提取能力
从前文的描述中,不难发现,深度学习并不是一种新的发明。它是基于ANN发展而来的,而ANN的数学模型早在1943年就被McCulloch和Pitts开发出来。直到1986年,Hinton和他的合作者将BP算法融入ANN之后,ANN才变成了一个实用而且可训练的机器学习方法。随着计算资源的不断发展,含有多隐层的深度神经网络在最近十多年开始变得流行,主要是因为先前提到的一些训练算法的发展以及计算能力的提高使得深度网络架构的潜在能力得以展现,从而表现出强大的竞争力。因此深度学习的崛起并不仅仅是ANN的重新利用,更多的是计算能力的快速发展以及训练技术的有效开发。事实上深度学习技术的应用一般来说都需要较大的数据量作为前提。由于化学分子数量多、结构复杂, 使用传统的算法处理信息时,能力常有不足,而使用深度学习技术有望突破这一局面[18]。
随着化学研究的不断发展,目前已经开发很多的分子描述符来描述化合物大量的性质。这些描述符通过化学知识和直觉被设计出来,将其作为特征输入传统的机器学习模型中。这些模型在化学信息学领域已经取得了较为瞩目的成绩[5,19-22]。在传统的机器学习算法中,线性回归(linear regression)、决策树(decision tree)算法构建的模型是比较直观且容易被理解的简单模型。对于有着非线性关系的复杂性质的预测,通常会采取更加复杂的机器学习算法构建一个具有较好预测能力的模型,如支持向量机、随机森林。
对于深度学习来说,它是一种用来预测非线性的复杂性质的算法,但是它与SVM和RF有较大的区别。其区别主要在于深度学习是将原始的输入信息进行转换,然后通过隐层的神经元将其重组成分布式的表征,配合一些合理的训练方式,每一层隐层的神经元能够通过无监督或者有监督的学习,提取抽象、分层以及深度的特征。这个过程被称作“自动特征提取”。在不需要领域知识和人工干预的情况下,自动提取特征是深度学习算法最重要的优点之一[23],这一点与传统机器学习算法完全不同。传统的机器学习算法构建的模型的优劣完全依赖于输入特征的好坏,所以对于传统机器学习模型来说,特征设计和选择是一项比较繁琐并且依赖于化学知识和直觉的工作。深度学习凭借自动的特征提取能力,使其在面对大量的标签和无标签数据时,不需要人工干预就能够自动学习到有用的特征信息,基于这些特征信息进行较好的决策,减轻了大数据时代特征工程的“烦恼”。如今,深度学习凭借特征自动学习能力已成为语音识别[1,24-27]、自然语言处理[3,28-30]、计算机视觉[2,8,31,32]的主流算法。在深度学习没有出现之前,最先进的模型在ImageNet的比赛上错误率为25%~30%,而识别人类图像的理想错误率是5.1%[33],所以当时的模型与人类的识别水平存在着较大的差距。2012年以后,深度学习算法被Hinton等人引入计算机视觉领域,并应用到了ImageNet的比赛中,其错误率被显著降低到16.4%[2]。随后在2015年,微软亚洲研究院(Microsoft Research,MSRA)的团队利用深层的网络框架打破了人类的识别水平,将错误率降至4.95%[31]。2016年,来自MSRA的深度残差网络将图像识别的错误率降低到3.57%[34],这充分说明深度学习所具备的潜力。不同的深层网络架构赋予了模型不同的潜能,不同的训练技术更是让模型展现出惊人的表现。而这一切都归因于深度学习的崛起,大数据的发展以及计算能力的进步铸就了深度学习的成功。在大量的数据面前展现出的自动提取特征的能力让深度学习驰骋于大数据时代的战场。深度学习并不仅仅促进了计算机视觉、自然语言处理和语音识别领域的飞速发展,也促进了其他领域的快速发展,比如化学信息学、生物信息学等。
3 深度学习在QSAR中的应用
传统的机器学习算法在化学信息学领域的应用已经有了很长的历史,其中最值得关注的是QSAR的应用。在训练集已知的前提下,通常一个QSAR模型的构建会涉及以下两个过程。
● 编码过程:将一个化合物转化成有效的表征。通过化学专业知识设计出描述化合物性质的分子描述符,从而计算出用来表征化合物性质或者拓扑结构的x,即模型的输入特征。
● 映射过程:即模型构建过程,发现一个函数f使得输入特征x与目标性质y之间产生经验性的联系,即y≈f(x)。
早期的QSAR模型先是基于线性回归模型和贝叶斯神经网络,随后是RF和SVM。但是这些模型都依赖于特征的设计和选择。而近年来发展的基于深度学习的QSAR模型也逐渐进入研究人员的视线。下面就基于模型框架的类型分别讨论近年来深度学习技术在QSAR中的进展情况。
3.1 深层神经网络框架
DNN框架是最传统的一套深度学习框架,在QSAR中,该模型的能力在一次次应用和比赛中被推向了顶峰。
Dahl和Hinton带领的团队在2012年首次将深度学习引进QSAR中,并赢得了Merck赞助的Kaggle比赛。在15个药物靶标的预测比赛中,DNN模型的平均R2(皮尔森相关系数平方)以0.494的成绩打败了RF模型(0.420)[35],虽然这只是一个很微弱的优势,但是是近十多年中首次打破RF模型领军地位的模型。该方法在数据的编码过程采用了“atom pair”描述符(来自Carhart等人[36])和“donor-acceptor pair”描述符(来自Kearsley等人[37])。其描述符的基本范式是:原子i—(键距)—原子j。在映射过程中,该方法建立在一个简单的DNN框架上,加上了避免梯度消失的ReLU激活函数,采取了先前提到的dropout和无监督预训练的训练策略,然后利用批量随机梯度下降法[38]对模型进行训练以及优化,从而建立了药物和靶标之间的映射关系。该方法后来被Merck团队在扩充的数据集上进行综合性的评估和分析,他们发现该DNN方法能够在很多任务上轻松地超过RF模型,并强调了该方法可以作为一种实用的开发QSAR模型的技术[39]。
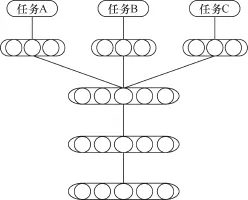
图5 多任务模型神经网络框架示意
受到先前深度学习表现的鼓舞之后,Dahl 等人[40]在2014年将单任务的基于DNN的QSAR模型发展为多任务的基于DNN的QSAR模型,主要原因是多任务学习(如图5所示)所具备的一些优势:能够产生一套更加普适的特征;权值被更多的数据优化以及被共享;一个模型能够解决多个任务;有可能提高模型的表现等[41]。该方法被应用于PubChem的19套芯片数据中。其编码方式是利用Dragon计算的3 764个Dragon分子描述符[42],每一个描述符用Z-score进行转化。在映射过程中,该方法采用了先前的ReLU、dropout和无监督预训练的训练策略,加入了贝叶斯优化算法来优化超参数[43,44],在批量随机梯度下降时,为了防止目标函数损失发生偏差,每个小批量中的样本由来自每套芯片数据的训练样本组成。例如,要建立一个7任务的模型,在训练时每个小批量有80个样本,其中20个数据来自需要更加关注的芯片数据,还有60个数据来自其他6组芯片的每组10个随机样本。通过上述方式开发基于DNN的多任务QSAR模型。在二分类问题上,该多任务的深度学习模型比传统的机器学习算法(RF、梯度提升树[45])在12组芯片上有了显著的提高,而在其他7组芯片上也达到了较为相近的水平。该方法的提出同时也掀起了多任务QSAR模型的新篇章。
2014年,Hochreiter等人[46]在将多任务的DNN方法扩展到了一个更大的数据集——大规模的具有生物活性的化合物数据集ChEMBL[47]。该数据集包含了1 300万个ECFP12分子描述符[48]、130万个化合物、5 000个药物靶标,数据量显著高于Kaggle 2012数据集(包含11 000个分子描述符、164 000个化合物、15个药物靶标)。Hochreiter等人利用ECFP12的编码方式对数据集中化合物的子结构进行编码,组成了一个超高维的稀疏矩阵,预处理之后得到了700 000(compounds)×43 340(输入特征)的矩阵作为DNN模型框架的输入。在映射过程中,采用未公开的训练策略,构建了超大规模的多任务DNN模型,能够使得1 230个靶标同时进行预测,并将之与传统的机器学习算法(如SVM、Binary Kernel Discrimination[49]、Logistic回归、k近邻)以及一些商业软件的方法(如基于ParzenRosenblatt KDE的方法[50]、基于Pipeline Pilot的贝叶斯分类方法[51]、Similarity Ensemble Approach[52])进行比较。他们用实验证明了多任务的DNN模型表现出来的能力比其他传统机器学习方法都要略高一筹。多任务DNN模型在准确率上达到了0.830的ROC曲线的线下面积(area under curve,AUC),其他模型的准确率最高也只有0.816 AUC。与2014年Dahl 等人对多任务的评估结论保持一致,充分肯定了多任务DNN模型在QSAR中具备的优势。在模型的预测结果中,Hochreiter等人还注意到在单任务中数据量小,难以训练的模型,在多任务中能够很好地完成训练,同时也能有不错的表现。这是由于多任务学习涵盖了迁移学习的概念,将其他不同但相关的任务学习到的有效特征转移到了具有少量数据集的表征中,使得模型在面对少量数据集时仍然保持令人满意的表现。与此同时,多任务DNN模型隐层中对于化合物分层、抽象、复杂的表征,有潜力被迁移至其他的相关任务中。
Ramsundar等人在2015年整合了PubChem的小分子生物活性的芯片数据(PubChem’s bioassay database,PCBA)[53]、基于PCBA发展的用于虚拟筛选的最大无偏验证(maximum unbiased validation,MUV)数据集[54]、用于分子对接评估的假阳性化合物库(增强版)(directory of useful decoysenhanced,DUD-E)数据集[55]以及NIH组织毒性预测比赛的Tox21数据集。最终收集到了259个药物靶标,包括160万个化合物,3 780万个实验测量数据点[56]。利用扩展连通性指纹(extended-connectivity fingerprints,ECFP4)的分子片段编码方式[48]对化合物进行编码,随后将其输入多任务DNN框架中。Ramsundar等人也采用了ReLU和Dropout的训练策略构建多任务QSAR模型。并将该模型与传统的机器学习模型(Logistic回归模型、RF模型)进行比较,发现多任务的DNN模型的表现在PCBA(0.873 AUC)、MUV(0.841 AUC)、Tox21(0.818 AUC)3个数据集上要明显优于传统机器学习模型。同时作者还设计了多任务和单任务的比较实验,从实验的角度说明了数据量一定时,随着任务数量的增加,模型的表现也会变得越好,同时也发现当任务数一定时,数据量越大,模型也会有更好的表现。除了与先前的多任务DNN模型[46,48]保持一致的结论外,在实验中Ramsundar等人还发现了多任务DNN模型的迁移能力是受到一定限制的。它既受到模型的初始化限制,也受到是否与训练集相关的限制。
Hochreiter等人[57]在2016年再一次利用多任务的DNN框架开发化合物毒性评估模型DeepTox,并且在NIH组织的Tox21的比赛中取得了优异的成绩。该数据集有12 000个化合物,对应着12个芯片的毒性测量数据。DeepTox模型事实上沿用了2014年他们开发的多任务DNN框架[57],并采取了相似的训练手段(ReLU 和Dropout),唯一不同的是编码的方式采用了静态编码(3D和2D的分子描述符及预先定义的2 500个毒性基团[58]等)和动态编码(ECFP、DFS、RAD2D等)。然后将其输入一个含有多隐层的DNN中进行训练,从而开发出DeepTox模型。多任务的DNN模型DeepTox在面对12组芯片数据的测试集的挑战时,有9组芯片的测试结果优于单任务的模型。该模型相比其他传统的机器学习模型(SVM、RF、ELNet[59,60]),DNN模型以平均0.837 AUC的优异表现略高一筹。除了对模型的预测能力进行评估之外,Hochreiter等人还设计了对隐层的可视化实验,利用ECFP编码去除2 500个毒性片段作为模型的输入,从而开发模型,发现隐层的神经元的激活度与毒性基团有着较为明显的联系(如图6所示)。这种对隐层特征的探索,直接印证了深度学习的特征组合能力,使得开发基于深度学习的化合物生成模型变成了可能。
以上是基于DNN框架的QSAR模型的简单介绍。该架构比较直观和简单,但是该类模型的表现能力伴随着深度学习训练技术和计算能力的发展、数据集的扩充而不断开创佳绩。
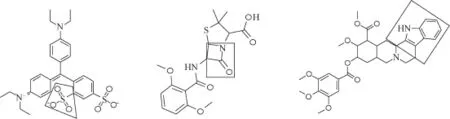
图6 Hochreiter等人设计的实验中隐层中与毒性相关的片段(框中部分为毒性片段)
3.2 卷积神经网络框架
传统DNN框架在QSA R模型中的实现已经在第3.1节中介绍。而在计算机视觉领域风靡的CNN框架对QSAR模型的开发更加智能化,在信息输入时,只需要最原始的简化分子线性输入规范(simplified molecular input line entry specification,SMILES)便能进行模型的训练工作,避免了不少特征工程的工作。
Duvenaud等人[61]利用CNN编码分子图,获得小分子分布式的表征,并通过模型训练和优化构建端对端的QSAR模型,能够直接从SMILES映射到性质,开发了一套基于神经网络的分子指纹(neural fingerprint,NFP)的方法。前文提到的所有QSAR模型构建的基本范式是先用特征计算软件计算化合物分子的多种描述符,然后将其作为模型的输入从而开发模型。而NFP是基于分子图卷积的方式,能够直接从化合物的SMILES格式出发,将其转化为2D的图结构,然后通过CNN将不定大小的分子图表征为定长的向量,随后将其输入全连接的ANN中,通过有监督学习对模型进行训练,从而构建QSAR模型,整个过程中没有参与任何的人工干预,成为了智能化的QSAR。该方法是受到神经图灵机[62]思想的启发,将Morgan算法[63]编码分子离散的3部分——“Hash”“Index”“Write”替代为“CNN”“Softmax”“Add”,使整个模型变成一个连续可微的系统。图7展示了CNN是如何编码分子图的。类似于ECFP的迭代提取子结构,这里是将每一步迭代过程中的信息传递用CNN进行转移,然后从每一个迭代层中提取信息,再把这些信息累加起来,作为该分子的表征或者指纹,将其输入标准的ANN中,实现端对端的可微系统。其中模型的输入信息来自原子的特征描述和原子间键的特征描述。在训练过程中超参数的优化、自动微分工具包Autograd、梯度优化技术Adam被用来训练和优化模型。该方法被应用到了3个数据集中:对于水溶性预测[64],该方法达到了0.52±0.07 均方根误差(RMSE)的水平;在药物有效性的预测[65]中,该方法达到了1.16±0.03 RMSE的水平;在有机光伏转化的预测[66]中,该方法达到了1.43±0.09 RMSE的水平。这些数据集在该方法中的成功应用说明了该方法具有较好的普适性。比较有趣的是该方法中隐层的抽象表征并非像其他深度神经网络一样无法解释,这些隐层表征与ECFP描述符存在着较高的相似性,同时该表征能够与目标性质相关的片段建立起较强的联系(如图8所示),能够使模型学出与目标性质相关的特征,直接地展现了深度学习的特征自动提取能力,有助于开发智能化的化合物生成模型。
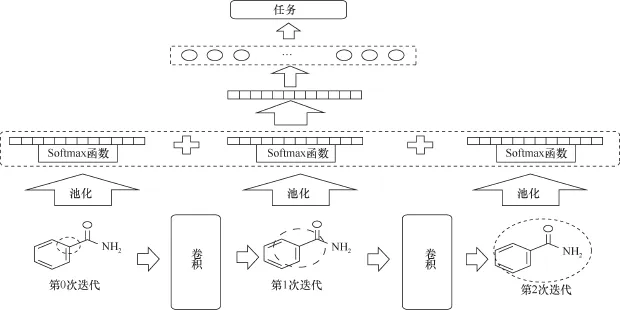
图7 基于Morgan算法的分子图卷积的框架示意
图8 Duvenaud等人方法中隐层中与目标性质相关的分子片段(框中部分为学到的分子片段)
随后Kearnes等人[67]基于类似的想法开发了另外一套基于分子图卷积(molecular graph convolution,MGC)的方法,将原子水平的特征(原子特征及原子之间键的特征)依据图的拓扑结构,利用4个操作(原子→原子(A→A)、键→键(P→P)、键→原子(P→A)、原子→键(A→P))通过CNN对每个中心原子进行特征提取,从而构建分子水平的表征,然后通过BP算法优化这些表征,从而完成多任务模型的开发(如图9所示)。在训练过程中,对多个数据集进行评估后,在PCBA数据集上的中值AUC可以达到0.909的水平,在MUV数据集上是0.875 AUC,在Tox21数据集上是0.867 AUC。Kearnes等人还将其与传统的机器学习方法(Logistic回归、RF)以及先前提到的基于分子描述符的DNN的方法进行比较,发现他们的MGC方法在前面提到的3个数据集上的表现都优于Logistic回归、RF和基于分子描述符的DNN方法。图卷积方法在多任务模型上取得较为明显的优势后,Kearnes等人将其与Duvenaud等人的方法数据集进行比较,水溶性、药物有效性和有机光伏转化性预测上分别以0.06、0.09、0.33 RMSE的微弱优势领先,但是值得注意的是,图卷积的方法是直接将这3个数据集进行多任务的训练,而Duvenaud等人是对每个数据集进行单任务的训练,所以这样的比较并不公平。这套智能化的MGC方法在多任务的预测上所展现出的实力使Kearnes等人期望MGC方法在未来的应用中能够超越所有基于分子指纹或者描述符的方法。然而,虽然该方法在模型的表现上有着一定的优势,但是对于模型的解释仍然是一个“黑盒”,并没有像NFP方法一样具有可解释性,一个可解释的模型才是化学家比较认可的模型。
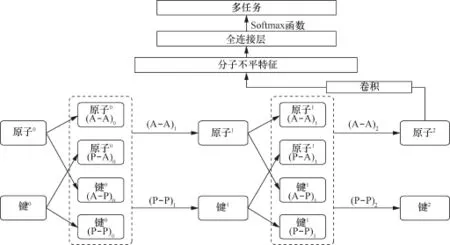
图9 Kearnes等人所用的模型框架示意
这两种基于图卷积的分子编码方式的主要思想是将原子水平的信息作为输入,依据分子图的拓扑结构利用深度学习将其组合为分子水平的信息,避免了先前直接从分子水平计算特征而造成的信息压缩或噪音,同时也避免了直接利用原子水平特征输入DNN导致的各个神经元之间信息的盲目组合。

图10 Swamidass等人设计的基于CNN的框架示意
前文提到的两个CNN框架都是用来预测分子层面的性质。Swamidass等人[68]将CNN应用到类药小分子环氧化作用的机理中,从而判断类药分子的毒性。Swamidass等人设计了专门针对该类问题的深度学习架构,如图10所示。该架构包含1个输入层、2个隐层、2个输出层。在编码过程中,先构建了原子间键的编码(左原子—键—右原子),然后计算了分子水平的描述符。模型的训练分为两个阶段:首先训练原子水平的网络模型,从而预测该化学键是环氧化位点的可能性;然后将前面模型的输出以及分子描述符作为分子水平网络的输入,通过训练构建分子水平的模型,判断该分子是否为环氧化分子。Swamidass等人利用该网络架构训练了702个环氧化反应,在环氧化位点识别上表现出0.949 AUC,在区分环氧化分子上表现出0.793 AUC。这是深度学习在机理模型研究上的首次成功应用,该模型不仅能够预测类药分子的环氧化作用,还能预测分子中的环氧化作用位点。该研究组还将类似的模型架构应用到了小分子与软亲核试剂的反应预测中,并对是否能够发生反应进行了预测,其准确率为80.6%,还预测了小分子的反应位点,其准确率为90.8%[69]。该机理模型对于数据集的标注比较繁琐,需要对化合物的每一个键进行性质的标注,并且要求训练数据集要清楚分子反应机理。
2015年Wallach等人[70]发表了基于3D结构的CNN模型——AtomNet,用来预测小分子和蛋白口袋的相互作用。该模型的输入信息是利用1埃间隔的3D格点在复合物的结合位点进行采样得到的。整个网络模型的架构与Krizhevsky等人[2]的深层CNN类似。利用多个3D的卷积核对输入信息进行特征提取,然后将提取的信息输入双隐层的全连接网络,最后进行决策。AtomNet采用ReLU和AdaDelta的训练策略来优化模型。该模型在4个测试集上获得了0.745~0.895的平均AUC,显著优于Smina[71](0.552~0.700 AUC)。同时与前人的结果进行较为公平的比较,发现AtomNet相比以前的对接方法有着显著的提高,例如,Gabel 等人[72]使用Surflex-Dock[73]在10个DUDE的靶标上得到中值AUC为0.760,而AtomNet为0.930 AUC;Coleman 等人[74]使用DOCK3.7 在DUDE的所有靶标上得到平均AUC为0.696,而AtomNet为0.895 AUC;Allen 等人[75]使用Dock6.7在5个DUDE的靶标上得到平均AUC为0.72,AtomNet为0.852 AUC。Pereira 等人[76]也利用类似于在自然语言处理中比较流行的word embedding的方法构建了CNN的框架,预测小分子和蛋白口袋的相互作用。其发展的DeepVS-ADV模型在DUD的40个靶标中表现出0.810的平均AUC,为化学信息学提供了一套新的分布式的分子表征方法。
以上是CNN在QSAR模型中的应用进展情况介绍。其中Duvenaud等人和Kearnes等人的方法为化合物的性质预测提供了一套智能化的QSAR模型构建方式,并拥有高水平的预测能力,加快了基于性质预测的QSAR模型的开发。Wallach等人和Pereira等人提出了基于深度学习的3D结构表征方式,促进了化学信息学的发展。
3.3 循环或递归神经网络框架
Lusci等人[77]在2013年利用无向图递归神经网络(UGRNN)预测类药小分子的水溶性,如图11所示。首先将小分子的2D结构看成一个由原子和键组成的无向图,定义每个重原子为无向图的根节点,使无向图转化为树结构,然后所有的其他节点向该节点沿着最短路径进行汇聚,原子之间的信息用一个3层的神经网络进行传递,由根节点生成定长的向量F表征这棵树。一个无向图有N个重原子,就有N棵类似的树结构,也就有N个类似的定长向量,将这些定长的向量相加得到了分子水平的表征向量F,然后加入全连接的网络进行决策,如图11所示。Lusci等人通过BP算法更新共享的权值,从而训练整个网络,最后通过组合模型策略对输出结果进行评估。该模型对水溶性的3个数据集的表现分别为0.92 R2,0.91 R2,0.81 R2,优于先前的其他一些模型[78-81]。笔者团队也对该方法进行了尝试,利用UGRNN预测了类药分子的较为复杂的一类性质——肝毒性(药物引起的肝损伤)。该方法在多个数据集上都表现出较为不错的预测能力[82]。笔者在模型训练中发现该方法的计算复杂度O(|F|2N2)比较高,导致模型收敛速度特别慢。该方法与先前提到的图卷积法一样,不能对隐层学到的特征进行较为合理的解释。
LSTM是RNN中应用最广泛、最成功的模型。Pande课题组将LSTM应用于多任务QSAR中,基于one-shot学习[83]开发了ResLSTM(Residual LSTM)方法[84]。该方法被证明是一套较为不错的方法。比起传统的机器学习方法(RF),该方法在Tox21、SIDER[85]数据集的表现为0.757~0.840 AUC、0.602~0.752 AUC,而RF只有0.536~0.584AUC、0.501~0.546 AUC的水平。
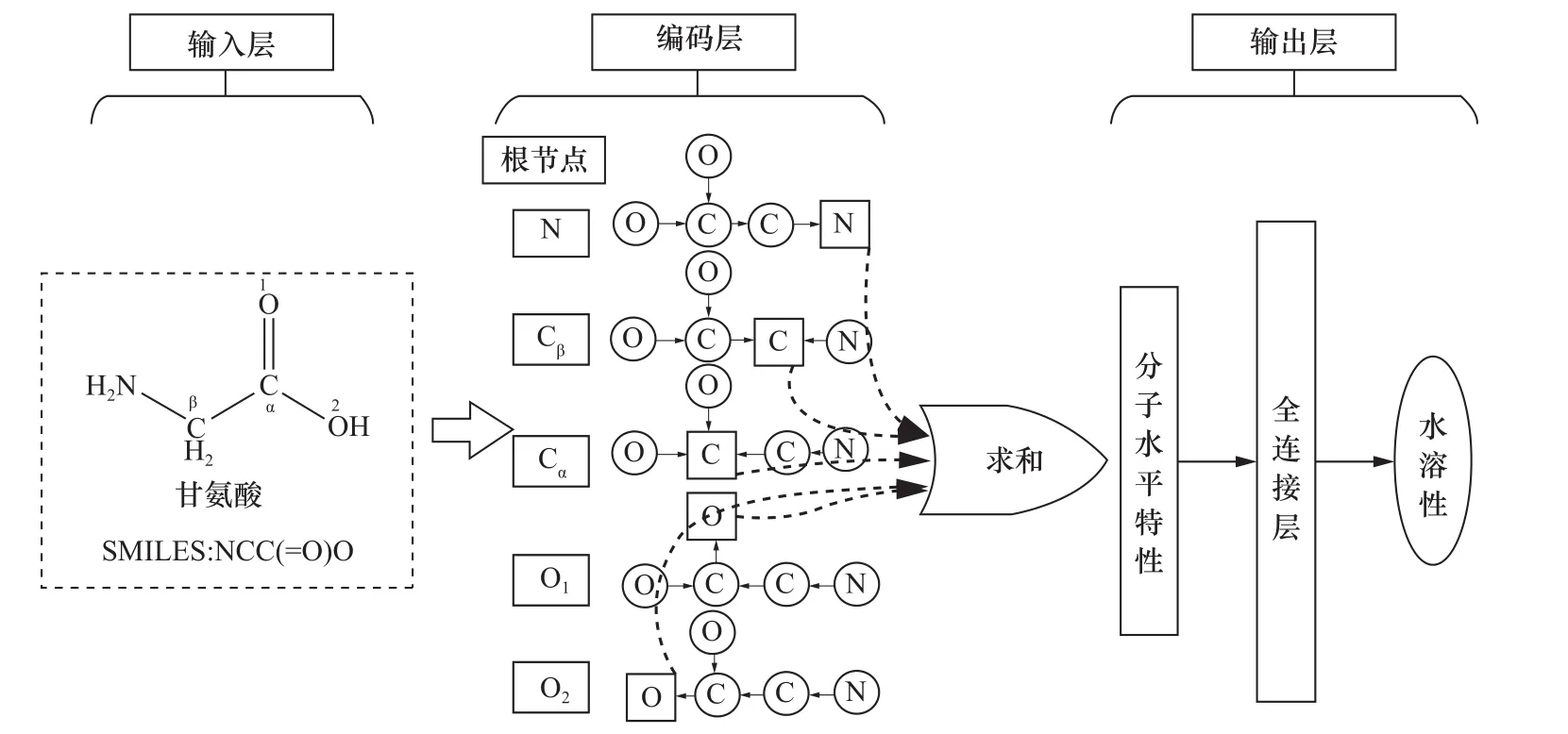
图11 UGRNN编码小分子的示意
总体来说,RNN在QSAR模型中的应用还比较少。但这并不能掩盖RNN在化学信息学中具备的潜在能力。近来RNN框架已应用于构建生成模型,从而设计与输入化合物性质相似的新的合理的小分子[86,87],充分体现了RNN在药物设计领域具有较好的应用前景。
4 深度学习框架的对比与分析
表1是深度神经网络框架在QSAR中的应用,可以看出,目前深度学习框架下的QSAR研究主要有以下几个特点。
● 随着数据集的增多以及多样化,研究人员逐渐倾向于使用多任务模型的训练策略,多任务学习中迁移学习的概念被应用到了数据较少的数据集中,提高对该任务的预测能力。多任务学习模型的评估方法大多是基于AUC的,说明多任务模型目前只适用于分类问题,在多任务的回归模型的问题上, 还有待开发出更好的训练手段和策略。
● ReLU目前是在QSAR中最常用的一种训练技术,在DNN和CNN框架中基本都使用了该技术。发展更好、更快的训练
技术有助于开发高效的QSAR模型。

表1 深度神经网络框架在QSAR 中的应用
从分子编码技术在深度学习中的应用来看,笔者发现基于原子水平的特征输入在逐渐取代基于分子描述符或指纹的特征输入,这说明深度学习拥有足够的能力从原子层面提取支持分子水平预测的信息,印证了其强大的特征提取能力。但目前比较不足的是对于这些深层特征的深层分析。目前研究人员主要采用的策略是重新设计实验,专门用来可视化隐层中与目标性质相关的分子片段,并没有直接从构建出来的高水平的QSAR模型本身出发进行隐层特征的分析,这方面的研究有待加强。
5 总结与展望
综上所述,由于化学分子数量多、结构复杂多样,使用传统的算法处理时能力常有不足,深度学习的表现比起传统机器学习算法更胜一筹,主要是因为深度学习是一种多层描述的表征学习,通过组合简单、非线性模块来实现,每个模块都会将最简单的描述(从原始或近原始输入开始)转变成较高层、较为抽象的描述。其关键之处在于这些抽象的特征并非人工设计,而是模型从大量数据中自动学习得到的。这样的能力在面对化学中的大量实验数据时显得更为得心应手,更加智能化。从目前的应用表现来看,虽然深度学习在语音处理、计算机视觉和自然语言处理中的应用已经非常广泛,但是深度学习在QSAR乃至化学信息学中的应用目前还只属于初步的阶段。而这些应用表现出来的成功之处可以折射出深度学习在化学领域的应用前景中必然是一条康庄大道。从QSAR问题的复杂度来看,多任务QSAR模型的开发本来是一件很难完成的事情,然而在深度学习面前就显得相对简单,在模型表现上也显得极为突出。在QSAR模型编码时,初步发现一些依靠化学专业知识设计的特征(如分子描述符)已经不再那么重要,仅仅依靠非常简单的原子层面的信息就能组建高水平的QSAR模型。这无疑是归功于深度学习的强大特征学习能力。而且这些特征甚至可以在隐层中被转化为一些真实的化合物子片段的概念,如DeepTox中涉及的毒性片段以及NGF方法涉及的与目标性质相关的片段,促进了深度学习在QSAR中的可解释性的研究。深度神经网络是一套适合做“感知”的框架,让适合做“感知”的深度学习结合以推理为核心的贝叶斯神经网络,形成“感知—推理—决策”的范式,从而加快基于深度学习的新型药物设计的发展。
深度学习应用于化学信息学还存在一些需要解决的关键科学问题,包括如何进一步改进过拟合现象和加快深度神经网络的训练过程;如何发展更适用于分子二维及三维结构信息特征的编码方法和网络结构、超参数优化算法及多目标深度学习算法;如何准确预测化合物与生物网络的作用关系及其生物活性。如何高速有效地处理非结构化的化学分子相关文本文献和图像信息数据,也是一个需要解决的关键问题。深度学习对数据的强大处理和理解能力,也为人们提供了一条可能的新途径,以便更好地理解化学分子结构的物理化学本质。
[1]HINTON G, DENG L, YU D, et al. Deep neural networks for acoustic modeling in speech recognition: the shared views of four research groups[J]. IEEE Signal Processing Magazine, 2012, 29(6): 82-97.
[2]K RIZ HEVSKY A, SUTSKEVER I,HINTON G E. Imagenet classification with deep convolutional neural networks[J]. Advances in Neural Information Processing Systems, 2012, 25(2): 1097-1105.
[3]COLLOBERT R, WESTON J. A unified architecture for natural language processing:deep neural networks with multitask learning[C]// The 25th International Conference on Machine Learning, July 5-9, 2008, Helsinki, Finland. New York: ACM Press, 2008: 160-167.
[4]GAWEHN E, HISS J A, SCHNEIDER G. Deep learning in drug discovery[J]. Molecular Informatics, 2016, 35(1): 3-14. [5]RAGHU M, POOLE B, KLEINBERG J, et al. On the expressive power of deep neural networks[J]. Statistics, 2016, arXiv:1606.05336.
[6]HINTON G E, OSINDERO S, TEH Y W. A fast learning algorithm for deep belief nets[J]. Neural Computation, 2006, 18(7):1527-1554.
[7]S R I VA S TAVA N, H I N T O N G E, KRIZHEVSKY A, et al. Dropout: a simple way to prevent neural networks from overfitting[J]. Journal of Machine Learning Research, 2014, 15(1): 1929-1958.
[8]I O F F E S, S Z E G E D Y C. B a t c h normalization: accelerating deep network training by reducing internal covariate shift[J]. Computer Science, 2015, arXiv:1502.03167.
[9]GLOROT X, BORDES A, BENGIO Y. Deep sparse rectifier neural networks[C]// The 14th International Conference on Artificial Intelligence and Statistics, April 11-13, 2011, Fort Lauderdale, USA. [S.l.:s.n.], 2011: 315-323.
[10]DUCHI J, HAZAN E, SINGER Y. Adaptive subgradient methods for online learning and stochastic optimization[J]. Journal of Machine Learning Research, 2011, 12(7): 2121-2159.
[11]ZEILER M D. ADADELTA: an adaptive learning rate method[J]. Computer Science, 2012, arXiv: 1212.5701.
[12]KINGMA D, BA J. Adam: a method for stochastic optimization[J]. Computer Science, 2014: arXiv: 1412.6980.
[13]MIKOLOV T, KARAFIÁT M, BURGET L, et al. Recurrent neural network based language model[C]//The 11th Annual Conference of the International Speech Communication Association, September 26-30, 2010, Makuhari, Chiba. [S.l.:s.n.], 2010: 1045-1048.
[14]WU Y, SCHUSTER M, CHEN Z, et al. Google's neural machine translation system: bridging the gap between human and machine translation[J]. Computer Science, 2016, arXiv: 1609.08144.
[15]VINCENT P, LAROCHELLE H, LAJOIE I, et al. Stacked denoising autoencoders:learning useful representations in a deep network with a local denoising criterion[J]. Journal of Machine Learning Research, 2010, 11(12): 3371-3408.
[16]SOCHER R. Recursive deep learning for natural language processing and computer vision[J]. Citeseer, 2014(8): 1.
[17]HOCHREITER S, SCHMIDHUBER J. Long short-term memory[J]. Neural Computation, 1997, 9(8): 1735-1780.
[18]孙潭霖, 裴剑锋. 大数据时代的药物设计与药物信息[J]. 科学通报, 2015(8): 689-693. SUN T L,PEI J F. Drug design and drug information is the big data era[J]. Chinese Science Bulletin, 2015(8): 689-693.
[19]SVETNIK V, LIAW A, TONG C, et al. Random forest: a classification and regression tool for compound classification and QSAR modeling[J]. Journal of Chemical Information and Computer Sciences, 2003, 43(6): 1947-1958.
[20]RUPP M, TKATCHENKO A, MÜLLER K R, et al. Fast and accurate modeling of molecular atomization energies with machine learning[J]. Physical Review Letters, 2012, 108(5): 3125-3130.
[21]RACCUGLIA P, ELBERT K C, ADLER P D F, et al. Machine-learning-assisted materials discovery using failed experiments[J]. Nature, 2016, 533(7601): 73-76.
[22]DU H, WANG J, HU Z, et al. Prediction of fungicidal activities of rice blast disease based on least-squares support vector machines and project pursuit regression[J]. Journal of Agricultural and Food Chemistry, 2008, 56(22): 10785-10792.
[23]LECUN Y, BENGIO Y, HINTON G. Deeplearning[J]. Nature, 2015, 521(7553):436-444.
[24]JAITLY N, NGUYEN P, SENIOR A W, et al. Application of pretrained deep neural networks to large vocabulary spee ch re cog nition[C]//The 13th Annual Conference of the International Speech Communication Association, September 9-13, 2012, Portland, OR, USA. [S.l.:s.n.], 2012: 1-4.
[25]DAHL G E, YU D, DENG L, et al. Context-dependent pre-trained deep neural networks for large-vocabulary speech recognition[J]. IEEE Transactions on Audio, Speech, and Language Processing, 2012, 20(1): 30-42.
[26]GRAVES A, MOHAMED A R, HINTON G. Speech recognition with deep recurrent neural networks[C]//2013 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), May 26-31, 2013, Vancouver, BC, Canada. New Jersey: IEEE Press, 2013: 6645-6649.
[27]DENG L, YU D, DAHL G E. Deep belief network for large vocabulary continuous speech recognition: 8972253[P]. 2015-03-03. [28]GAO J, HE X, DENG L. Deep learning for web search and natural language processing[R]. Redmond: Microsoft Research, 2015.
[29]MIKOLOV T, SUTSKEVER I, CHEN K, et al. Distributed representations of words and phrases and their compositionality[J]. Advances in Neural Information Processing Systems, 2013, arXiv:1310.4546.
[30]SOCHER R, LIN C C, MANNING C, et al. Parsing natural scenes and natural language with recursive neural networks[C]//The 28th International Conference on Machine Learning (ICML-11), June 28-July 2, 2011, Bellevue, Washington, USA. [S.l.:s.n.], 2011:129-136.
[31]H E K, Z H A NG X, R EN S, et a l. Delving deep into rectifiers: surpassing human-level performance on imagenet classification[C]//The IEEE International C onference on C omputer Vision, December 13-16, 2015, Santiago, Chile. New Jersey: IEEE Press, 2015: 1026-1034. [32]SZEGEDY C, LIU W, JIA Y, et al. Going deeper with convolutions[C]//The IEEE Conference on Computer Vision and Pattern Recognition, June 7-12, 2015, Boston, MA, USA. New Jersey: IEEE Press, 2015: 1-9.
[33]RUSSAKOVSKY O, DENG J, SU H, et al. Imagenet large scale visual recognition challenge[J]. International Journal of Computer Vision, 2015, 115(3): 211-252.
[34]H E K, Z H A NG X, R EN S, et a l. Deep residual learning for image recognition[C]//The IEEE Conference on Computer Vision and Pattern Recognition, June 27-30, 2016, Las Vegas, NV, USA. New Jersey: IEEE Press, 2016: 770-778.
[35]MARKOFF J. Scientists see promise in deep-learning programs[N]. New York Times, 2012-10-25.
[36]C A R H A R T R E, S M I T H D H, VENKATARAGHAVAN R. Atom pairs as molecular features in structure-activity studies: definition and applications[J]. Journal of Chemical Information and Computer Sciences, 1985, 25(2): 64-73.
[37]KEARSLEY S K, SALLAMACK S, FLUDER E M, et al. Chemical similarity u s i n g p h y s i o c h e m i c a l p r o p e r t y descriptors[J]. Journal of Chemical Information and Computer Sciences, 1996, 36(1): 118-127.
[38]RUMELHART D E, HINTON G E, WILLIAMS R J. Learning representations by back-propagating errors[J]. Cognitive Modeling, 1988, 5(3): 1.
[39]MA J, SHERIDAN R P, LIAW A, et al. Deep neural nets as a method for quantitative structure-activity relationships[J]. Journal of Chemical Information and Modeling, 2015, 55(2): 263-274.
[40]DAHL G E, JAITLY N, SALAKHUTDINOV R. Multi-task neural networks for QSAR predictions[J]. Computer Science, 2014, arXiv: 1406.1231.
[41]EVGENIOU T, PONTIL M. Regularized multitask learning[C]//The 10th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, August 22 - 25, 2004, Seattle, WA, USA. New York: ACM Press, 2004: 109-117.
[42]MAURI A, CONSONNI V, PAVAN M, et al. Dragon software: an easy approach to molecular descriptor calculations[J]. Match, 2006, 56(2): 237-248.
[43]SNOEK J, LAROCHELLE H, ADAMS R P. Practical bayesian optimization of machine learning algorithms[J]. Advances in Neural Information Processing Systems, 2012, arXiv: 1206.2944.
[44] SNOEK J, SWERSKY K, ZEMEL R S, et al. Input warping for bayesian optimization of non-st ationar y functions[C]// International Conference on Machine Learning, June 21-26, 2014, Beijing, China. [S.l.:s.n.], 2014: 1674-1682.
[45]FRIEDMAN J H. Greedy function approximation: a gradient boosting machine[J]. Annals of Statistics, 2001, 29(5): 1189-1232.
[46]U N T E R T H I N E R T, M A Y R A, KLAMBAUER G, et al. Multi-task deep networks for drug target prediction[J]. Neural Information Processing System, 2014: 1-4.
[47]GAULTON A, BELLIS L J, BENTO A P, et al. ChEMBL: a large-scale bioactivity d at a b a s e fo r d r u g d i s c ove r y[J]. Nucleic Acids Research, 2012, 40(D1):D1100-D1107.
[48]ROGERS D, HAHN M. Extendedconnectivity fingerprints[J]. Journal of Chemical Information and Modeling, 2010, 50(5): 742-754.
[49]HARPER G, BRADSHAW J, GITTINS J C, et al. Prediction of biological activity for high-throughput screening using binary kernel discrimination[J]. Journal of Chemical Information and Computer Sciences, 2001, 41(5): 1295-1300.
[50]LOWE R, MUSSA H Y, NIGSCH F, et a l. P re d icti n g the me cha n ism of phospholipidosis[J]. Journal of Cheminformatics, 2012, 4(1): 2.
[51]XIA X, MALISKI E G, GALLANT P, et al. Classification of kinase inhibitors using a Bayesian model[J]. Journal of Medicinal Chemistry, 2004, 47(18): 4463-4470.
[52]KEISER M J, ROTH B L, ARMBRUSTER B N, et al. Relating protein pharmacology by ligand chemistry[J]. Nature Biotechnology, 2007, 25(2): 197-206.
[53]WANG Y, SUZEK T, ZHANG J, et al. PubChem bioassay: 2014 update[J]. Nucleic Acids Research, 2014, 42(Database Issue): 1075-1082.
[54]ROHRER S G, BAUMANN K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data[J]. Journal of Chemical Information and Modeling, 2009, 49(2):169-184.
[55]MYSINGER M M, CARCHIA M, IRWIN J J, et al. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking[J]. Journal of Medicinal Chemistry, 2012, 55(14): 6582-6594.
[56]RAMSUNDAR B, KEARNES S, RILEY P, et al. Massively multitask networks for drug discovery[J]. Computer Science, 2015, arXiv: 1502, 02072.
[57]M A Y R A, K L A M B A U E R G, UNTERTHINER T, et al. DeepTox:toxicity prediction using deep learning[J]. Frontiers in Environmental Science, 2016, 3(8): 80.
[58]KAZIUS J, MCGUIRE R, BURSI R. Derivation and validation of toxicophores for mutagenicity prediction[J]. Journal of Medicinal Chemistry, 2005, 48(1): 312-320.
[59]FRIEDMAN J, HASTIE T, TIBSHIRANI R. Regularization paths for generalized linear models via coordinate descent[J]. Journal of Statistical Software, 2010, 33(1): 1.
[60]SIMON N, FRIEDMAN J, HASTIE T, et al. Regularization paths for Cox’s proportional hazards model via coordinate descent[J]. Journal of Statistical Software, 2011, 39(5): 1.
[61]DUVENAUD D K, MACLAURIN D, IPARRAGUIRRE J, et al. Convolutional networks on graphs for learning molecular fingerprints[J]. Advances in Neural Information Processing Systems, 2015, arXiv: 1509.09292.
[62]GRAVES A, WAYNE G, DANIHELKA I. Neural turing machines[J]. Computer Science, 2014, arXiv: 1410.5401.
[63]MORGAN H L. The generation of aunique machine description for chemical structures-a technique developed at chemical abstracts service[J]. Journal of Chemical Documentation, 1965, 5(2):107-113.
[64]DELANEY J S. ESOL: estimating aqueous solubility directly from molecular structure[J]. Journal of Chemical Information and Computer Sciences, 2004, 44(3): 1000-1005.
[65]GAMO F-J, SANZ L M, VIDAL J, et al. Thousands of chemical starting points for antimalarial lead identification[J]. Nature, 2010, 465(7296): 305-310.
[66]HACHMANN J, OLIVARES-AMAYA R, ATAHAN-EVRENK S, et al. The Harvard clean energy project: largescale computational screening and design of organic photovoltaics on the world community grid[J]. The Journal of Physical Chemistry Letters, 2011, 2(17):2241-2251.
[67]KEARNES S, MCCLOSKEY K, BERNDL M, et al. Molecular graph convolutions:moving beyond fingerprints[J]. Journal of Computer-Aided Molecular Design, 2016, 30(8): 595-608.
[68]HUGHES T B, MILLER G P, SWAMIDASS S J. Modeling epoxidation of drug-like molecules with a deep machine learning network[J]. ACS Central Science, 2015, 1(4): 168-180.
[69]HUGHES T B, MILLER G P, SWAMIDASS S J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione[J]. Chemical Research in Toxicology, 2015, 28(4): 797-809.
[70]WALLACH I, DZAMBA M, HEIFETS A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery[J]. Mathematische Zeitschrift, 2015, arXiv: 1510.02855.
[71]KOES D R, BAUMGARTNER M P, CAMACHO C J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise[J]. Journal of Chemical Information and Modeling, 2013, 53(8): 1893-1904.
[72]GABEL J, DESAPHY J R M, ROGNAN D. Beware of machine learning-based scoring functions on the danger of developing black boxes[J]. Journal of Chemical Information and Modeling, 2014, 54(10): 2807-2815.
[73]SPITZER R, JAIN A N. Surflex-Dock:docking benchmarks and real-world application[J]. Journal of Computer-Aided Molecular Design, 2012, 26(6): 687-699.
[74]COLEMAN R G, STERLING T, WEISS D R. SAMPL4 & DOCK3. 7: lessons for automated docking procedures[J]. Journal of Computer-Aided Molecular Design, 2014, 28(3): 201-209.
[75]ALLEN W J, BALIUS T E, MUKHERJEE S, et al. DOCK 6: impact of new features and current docking performance[J]. Journal of Computational Chemistry, 2015, 36(15):1132-1156.
[76]PEREIRA J C, CAFFARENA E R, SANTOS C N D. Boosting docking-based virtual screening with deep learning[J]. Journal of Chemical Information and Modeling, 2016, arXiv:1608.04844.
[77]LUSCI A, POLLASTRI G, BALDI P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules[J]. Journal of Chemical Information and Modeling, 2013, 53(7):1563-1575.
[78]JAIN N, YALKOWSKY S H. Estimation of the aqueous solubility I: application to organic nonelectrolytes[J]. Journal of Pharmaceutical Sciences, 2001, 90(2): 234-252.
[79]LOUIS B, AGRAWAL V K, KHADIKAR P V. Prediction of intrinsic solubility of generic drugs using MLR, ANN and SVM analyses[J]. European Journal of Medicinal Chemistry, 2010, 45(9): 4018-4025.
[80]A Z E N C O T T C A, K S I K E S A, SWAMIDASS S J, et al. One-to fourdimensional kernels for virtual screening and the prediction of physical, chemical, and biological properties[J]. Journal of Chemical Information and Modeling, 2007, 47(3): 965-974.
[81]FRÖHLICH H, WEGNER J K, ZELL A. Towards optimal descriptor subsetselection with support vector machines in classification and regression[J]. QSAR & Combinatorial Science, 2004, 23(5): 311-318.
[82]XU Y, DAI Z, CHEN F, et al. Deep learning for drug-induced liver injury[J]. Journal of Chemical Information and Modeling, 2015, 55(10): 2085-2093.
[83]LAKE B M, SALAKHUTDINOV R, TENENBAUM J B. Human-level concept learning through probabilistic program induction[J]. Science, 2015, 350(6266): 1332-1338.
[84]ALTAE-TRAN H, RAMSUNDAR B, PAPPU A S, et al. Low data drug discovery with one-shot learning[J]. Computer Science,2016, arXiv: 1611.03199.
[85]KUHN M, LETUNIC I, JENSEN L J, et al. The SIDER database of drugs and side effects[J]. Nucleic Acids Research, 2015, 44(D1): D1075.
[86]GÓMEZ-BOMBARELLI R, DUVENAUD D, HERNÁNDEZ-LOBATO J M, et al. Automatic chemical design using a datadriven continuous representation of molecules[J]. Computer Science, 2016, arXiv:1610.02415.
[87]SEGLER M H S, KOGEJ T, TYRCHAN C, et al. Generating focussed molecule libraries for drug discovery with recurrent neural networks[J]. Computer Science, 2017, arXiv:1701.01329.
Deep learning for chemoinformatics
XU Youjun, PEI Jianfeng
Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China
Deep learning have been successfully used in computer vision, speech recognition and natural language processing, leading to the rapid development of artificial intelligence. The key technology of deep learning was also applied to chemoinformatics, speeding up the implementation of artificial intelligence in chemistry. As developing quantitative structure-activity relationship model is one of major tasks for chemoinformatics, the application of deep learning technology in QSAR research was focused. How three kinds of deep learning frameworks, namely, deep neural network, convolution neural network, and recurrent or recursive neural network were applied in QSAR was discussed. A perspective on the future impact of deep learning on chemoinformatics was given.
deep learning, artificial intelligence, quantitative structure-activity relationship, chemoinformatics
TP301
A
10.11959/j.issn.2096-0271.2017019

徐优俊(1990-),男,北京大学前沿交叉学科研究院博士生,主要研究方向为药物设计与药物信息。

裴剑锋(1975-), 男,博士,北京大学前沿交叉学科研究院特聘研究员,主要研究方向为药物设计与药物信息。
2017-02-15
裴剑峰,jfpei@pku.edu.cn
国家自然科学基金资助项目(No.21673010)
Foundation Item: The National Natural Science Foundation of China (No.21673010)
猜你喜欢
农村青少年科学探究(2020年5期)2020-08-18 02:20:52
中国生物医学工程学报(2019年6期)2019-07-16 07:52:40
人民珠江(2019年4期)2019-04-20 02:32:00
新民周刊(2018年8期)2018-03-02 15:45:54
饮食科学(2017年12期)2018-01-02 09:23:20
自动化学报(2016年3期)2016-08-23 12:02:56
电测与仪表(2016年5期)2016-04-22 01:13:46
少儿科学周刊·少年版(2015年1期)2015-07-07 21:57:30
计算机工程(2014年9期)2014-06-06 10:46:47
机械工程与自动化(2014年3期)2014-05-07 12:49:22
