
美国快速通道与突破性疗法模式的比较研究与借鉴
2017-04-14 05:39:26丁锦希李苏菊李鹏辉
上海医药 2017年5期
丁锦希+李苏菊+李鹏辉


摘 要 目的: 比较研究美国快速通道和突破性疗法模式,为我国完善药品审评审批制度提供参考经验。方法:对比分析快速通道与突破性疗法的设置背景、准入标准、资格认证流程、加速机制及实施绩效,总结两种模式的共同点及各自特点。结果:现阶段,我国设立了新药优先审评模式,审评逐步向具有临床价值的新药倾斜,但是对于临床试验阶段的研发指导尚不足。结论:建议借鉴美国快速通道和突破性疗法的审评理念,并在完善现有优先审评模式基础上,强化对临床试验阶段的研发指导,提高疗效突破创新药的可获得性。
关键詞 快速通道 突破性疗法 比较研究
中图分类号:R951 文献标识码:C 文章编号:1006-1533(2017)05-0052-05
Comparison and reference of Americas fast track and breakthrough therapy system
DING Jinxi*, LI Suju, LI Penghui
(China Pharmaceutical University, Nanjing 211198, China)
ABSTRACT Objective: To compare the Americas fast track and breakthrough therapy system in order to provide reference for the innovative drug review and approval system of China. Methods: The backgrounds, admittance criterions, qualification process, mechanism and implementation performance between the fast track and breakthrough therapy were compared and analyzed and their common grounds and respective characteristics were summarized. Results: Although the priority review pattern is established in China, which makes our country gradually lay emphasis on the clinical value of the drug, the guidance for the research and development of drugs at clinical trial is still insufficient. Conclusion: It is suggested that China should learn from American review concepts and strengthen the guidance for the research and development of drugs at clinical trial on the basis of the improvement of the existing priority review so as to improve the availability of innovative drugs with breakthrough efficacy.
KEY WORDS fast track system; breakthrough therapy system; comparative study
恶性肿瘤、传染病、心脑血管疾病等重大疾病类型严重威胁人类健康。此类疾病普遍缺乏有效的治疗手段,患者生存质量状况不佳,临床迫切需求具有显著疗效提高的创新药物。然而,此类疾病的治疗药物,尤其是疗效突破的新药一般具有研发难度大、周期长、成本高且失败风险高等特点。因此,发达国家大多建立了向具有突破性疗效药物倾斜的注册审批制度体系,将有限的审评资源用于此类药物的审评,促进具有突破性疗效新药的上市以缓解供需矛盾。目前中国的新药注册审批制度尚存在诸多不足之处,且药品注册审评积压严重,影响了审评资源的合理配置,进一步加剧中国创新药物的供需矛盾。
快速通道(fast track)和突破性疗法(breakthrough therapy)是美国先后设立的两种新药特殊审评模式,通过临床试验指导和滚动审评机制,加快恶性肿瘤、严重传染病、罕见病等疾病类型的具有突破性疗效药物的研发上市进程。两种模式既存在相似之处,又有各自鲜明的特点。
因此,本文将从制度背景、适用条件、加速机制等方面入手,全方位对比分析美国快速通道和突破性疗法模式,探究两种模式的制度特征及异同点,发掘可供中国药品注册审批制度进一步完善的借鉴之处。
1 制度设立背景
20世纪80年代,美国面临着严重的药品注册申请积压问题。为了加快临床急需新药的上市速度,美国于1992年先后设立了优先审评(priority review)和加速审批(accelerated approval)两种新药特殊审评模式[1]。这两种模式的实施,尤其是优先审评对提高FDA审评效率,消化注册申请积压发挥了巨大作用。短短5年内,美国药品审评周期平均值由26个月迅速降低至10个月[2]。
然而,优先审评和加速审批主要作用于药品的注册审评及上市后监管阶段,欠缺对新药临床研发阶段的扶持激励,而临床试验研究又是造成新药研发“投入大、周期长、风险高”的主要因素。因此,为了提高具有治疗优势新药的临床研发效率,加快其上市进程,美国于1997年建立了快速通道。该模式重点作用于新药临床试验阶段,旨在通过FDA的指导促进临床试验的高效及顺利进行,实现对新药研发上市全生命周期的激励政策全覆盖[3]。
突破性疗法模式的设立源于美国癌症研究之友组织(friends of cancer research, FOCR)所发出的倡议。该组织认为,在现有的药品审评制度体系下,用于治疗肿瘤疾病药物的上市进程仍过于缓慢[4]。因此,需要新的特殊审评模式来加速在临床早期显现出突出疗效改善的肿瘤用药的研发和上市进程。最终,美国国会于2012年7月通过了《FDA安全与创新法案》,正式确立突破性疗法模式[5]。突破性疗法模式脱胎于快速通道,两者有较大相似性,但突破性疗法模式指向性更明确(主要专注于肿瘤用药),且FDA的扶持指导力度将更强[6]。
2 准入标准
2.1 适用范围
快速通道和突破性疗法的主要适用范围均为临床开发难度较大,且已初步显露出明显治疗优势的新药研发项目。
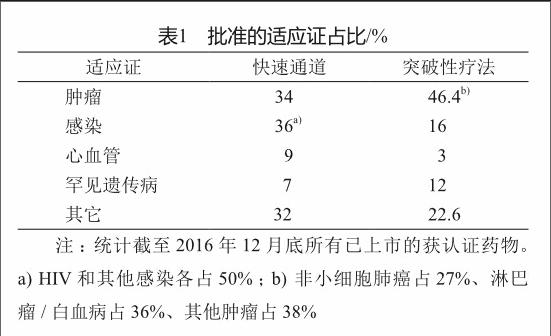
在实践中获得资格认证的药物均以新化学实体(NCE)药物和新生物制品为主;并主要集中于恶性肿瘤、严重病毒感染(HIV、丙肝等)及罕见遗传病等疾病类型。同时,这两种模式的认证药物类型和适应证在实施中有着较好的区分度。快速通道认证药物以感染、肿瘤、心血管疾病用药为主,对HIV等严重细菌/病毒感染的药物尤其重视;且认证药物大多为小分子化学NCE新药,占比高达60%,生物制品占比不足15%。突破性疗法认证药物以肿瘤药物为主(46%),对罕见遗传病药物的重视程度也较高(12%)(表1);同时,认证药物以生物制品为主体,占比45%,而NCE占比则为35%。
2.2 认证条件
美国《联邦法典第21篇》第356款和第506(a)款分别对快速通道和突破性疗法模式的资格认证条件做出明确规定。两种模式均适用于治疗严重危及生命疾病的新药,且有实质性的疗效改善,即能够解决未满足的临床需求。
但是,两种模式在证明药品有实质性疗效改善的证据方面存在明显差别。申请快速通道认证时,上述支持性证据的类型取决于药物研发所处的时期。在研发的早期,可运用非临床研究中的药物活性或药理学数据来证明这种潜力;在研发的较晚阶段,已有的临床数据应能证明该药疗效改善的潜力。突破性疗法则要求有初步的临床证据支持,认证条件更高。原因在于突破性疗法要求FDA给予药物临床试验更高的指导力度,资源及精力投入比快速通道更多。故只有在初期临床阶段显示出潜在优势的药物才能获得认证,以确保FDA有限的资源能被最优分配和利用。
3 资格认证流程
快速通道和突破性疗法遵循相同的资格认证流程(图1)。申请人需按规定格式及程序向FDA提交资格认证申请,FDA相关部门将在60 d内完成审查,并决定是否授予相应的资格认证。
两种模式由于认证条件中对支持性证据的要求存在差异,故申请提交的时机存在区别。快速通道使用非临床或临床支持证据均可,故常见的快速通道资格申请与IND申请同时提交。突破性疗法认证需有初步的临床证据,故FDA建议大多数的突破性疗法认证申请应作为IND的补充申请,在临床研发的早期阶段提交。
在收到资格认证申请后,FDA将组织审查人员重点审查,目前的证据能否支持该药获得相应的资格,及其临床研发计划能否证明药物具有疗效改善,可以解决未满足的临床需求。FDA将在60 d内完成审查并书面告知申请人,即发出资格确认函(designation letter)或否认函(non-designation letter)。此外,FDA可及时撤销不再符合快速通道或突破性疗法认证标准药物的资格认证。
4 加速机制
美国快速通道模式和突破性疗法模式均通过“临床试验指导”和“滚动审评”两种加速机制实现加快新药研发上市进程的制度目标。
4.1 临床试验指导
FDA认为新药临床试验计划的高效进行是快速通道至关重要的内容。FDA与申请人可通过固定交流协商会议及非固定的交流协商途径(电话、信函、邮件、非正式会晤等形式)实现对快速通道新药临床研究的及时介入指导,确保新药研发计划顺利制定及实施,并根据临床研究最新情况及时调整临床试验方案(图2)。
与快速通道相比,除上述临床试验指导途径外,突破性疗法新药还将获得FDA高层管理人员及有经验审评人员的密集介入交流,进一步强化指导力度。在上市审评过程中,审评小组可以成立专门的跨学科协作小组,来促进药物审评的高效进行。这样的跨学科协作小组将成为申请人和审批机构间的沟通渠道,在医学、临床药理学、毒理学等多方面为申请人提供建议和指导。
4.2 滚动审评
滚动审评(rolling review)指在申请人与FDA协商达成一致的前提下,申请人可分次提交药品注册申请材料,FDA将在收到申请人首次提交的材料后即进入药品上市审评程序,FDA边审评申请人边补充材料[7]。
一般而言,申请人会与FDA在前期协商中确定每次提交材料的时间和内容,申请人将按照这一时间表依次提交注册申请材料。各部分材料应格式完整,并能直接成为最终的NDA/BLA申请材料的一部分,例如完整的CMC(化學、生产与控制)部分、毒理学部分、临床试验部分等。
滚动审评机制使得FDA的审评和申请人准备注册申请材料的过程可以同时进行,审评更具灵活性。同时,申请人与FDA就滚动审评所进行的交流协商亦有助于确保注册申请材料的格式准确、证据充分,审评效率也将进一步提高。
5 实施绩效
截至2016年12月底,共计有80个药物研发项目获快速通道资格认证并已上市,45个药物获突破性疗法资格认证并已上市。
5.1 研发及审评周期
快速通道和突破性疗法模式均可大幅缩减新药研发周期。美国2016年上市的快速通道新药临床试验周期均值为6.9年,突破性疗法新药只有5.4年,较普通新药显著缩短。表明两种模式的“临床试验指导”机制在实践中发挥出良好的临床周期加速效果,且突破性疗法由于指导力度更大,临床周期进一步缩短(图3)。
此外,经快速通道或突破性疗法认证的新药,历年的注册审评周期均保持在6个月,短于普通新药10个月的审评周期(图4)[8]。由于快速通道及突破性疗法新药获得优先审评的几率更高,且“滚动审评”可进一步提高审评灵活性及效率,故其审评周期缩短。
5.2 认证及审批数量
近年来,FDA每年收到的快速通道资格认证申请约120个,认证率接近80%[9]。经过长期发展完善,快速通道已基本稳定,申请人及审评部门运用政策能力成熟,历年认证数量及认证率比较稳定。突破性疗法实施时间尚短,目前FDA每年接收到的突破性疗法认证申请总数约110个,历年认证率约30%[10]。表明突破性疗法的准入标准较快速通道更严格,以确保FDA资源的最优分配。
FDA历年通过快速通道审批上市药物数量有较大波动,一般不超过8个,占当年所有上市新药(NME或新生物制品的NDA/BLA申请)比例约5%。而突破性疗法尚处起步阶段,历年上市药物数量尚未趋稳(图5)。
由图5数据可见,两种模式历年的认证数量和上市数量间存在较大差距。例如,历年获快速通道资格认证的新药研发项目大约有80个,而年均上市的快速通道药物一般不超过8个,两者相差近十倍。原因可能是很多新药研发项目临床试验失败,从而最终无法上市;也可能是该项目随着研究进展不再符合资格认证标准,而被撤销了快速通道或突破性疗法的资格认证。
6 借鉴意义
目前,我国正在深化药品注册审评审批制度改革。2015年8月,国务院发布《关于改革药品医疗器械审评审批制度的意见》,为我国此轮药品注册审批制度改革指明了方向。国家食品药品监督管理总局亦积极探索,陆续发布一系列改革措施或征求意见稿,均意在提高仿制药质量,鼓励真正的创新研发活动。美国的快速通道与突破性疗法模式在实践中已表现出显著的激励扶持新药研发上市的制度效益,对我国药品注册审批制度改革,尤其是临床试验阶段的交流指导机制建设和完善具有重要的借鉴意义。
6.1 加强对突破性疗效创新药物的政策扶持力度
恶性肿瘤、心脑血管疾病等已经成为我国居民的主要致死疾病类型;我国的HIV、肝炎等重大传染病形势同样不容乐观;随着人口老龄化的加剧,各种慢性疾病和老年多发病的药物可获得性问题亦需引起重视[13]。然而我国医药产业仍以仿制为主,创新水平有限;同时药物注册积压问题严重,进一步妨碍了创新药物的上市进程。因此,我国亟待在政策层面强化对新药的激励扶持。
美国快速通道与突破性疗法模式的认证药物均以NCE和新生物制品为主体,同时亦包括有相当数量的新适应证、新剂型药物,充分体现出“疗效新”的认证理念。我国对新药的认识也逐渐从唯“结构新”转变为“疗效新”,CFDA于2016年2月26日发布的《关于解决药品注册申请积压实行优先审评审批的意见》,进一步强化了以疗效创新为新药的重要衡量标准。未来我国应持续完善注册审批制度,落实对具有突破性治疗优势新药的激励扶持政策,提高我国重大疾病领域的药物可获得性。
6.2 探索建立以临床试验指导为主要机制的新药特殊审评模式
美国先后共设立了四项新药特殊审评模式,通过不同的作用机制作用于新药研发上市全生命周期的不同阶段,更有针对性的指导新药研发[14]。其中,快速通道模式及突破性疗法模式主要作用于临床试验阶段,通过FDA的密集介入指导提高新药临床研发效率,进一步填补了以往特殊审评模式作用区域的空白。
目前我国已设立的优先审评审批制度主要作用于药物的临床试验申请和上市申请阶段,对药物临床试验阶段的指导力度明显不足。CFDA于2016年6月6日发布的《药物研发与技术审评沟通交流管理办法(试行)》初步建立完善了我国药品监管部门与申请人之间的沟通交流机制。未来我国可借鉴美国快速通道及突破性疗法模式,建立新的新药特殊审评模式,以临床试验指导为主要作用机制,进一步明确临床试验阶段的沟通会议。提高对突破性创新药物的临床试验指导扶持,实现对新药研发上市全过程的政策覆盖。
6.3 考虑引入滚动审评机制,提高审评灵活性和效率
美国获快速通道或突破性疗法模式认证的药物均适用滚动审评机制,滚动审评机制的实施提高了审评灵活性,一定程度上提高了审评效率。CFDA于2009年出台的《新药注册特殊审批管理规定》中规定申请人可以多途径“动态补充资料”——实行特殊审批的新药及首仿药可补充提交有利于药品资料完善的补充材料。但我国规定上市申请材料不能分次提交,故本质上不同于美国的滚动审评机制;同时这一规定可能导致注册申请资料质量不高,縱容申请人于审评过程中多次补充材料,严重影响审评审批效率。
因此,我国可借鉴美国滚动审评机制的成功经验,完善我国相关规定,并于适当时机引入滚动审评机制。强化申请人与监管部门在申请材料方面的交流协商,确保申请材料的格式准确,内容充分。通过允许分次提交上市申请材料,提高审评的灵活性及效率,加快临床急需药物的上市进程。
参考文献
[1] 杨莉, 连桂玉, 邢花, 等. FDA在新药注册审批中的研发激励机制研究[J]. 中国新药杂志, 2012, 21(9): 964-968.
[2] Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics [EB/OL]. (2016-12-20). http://www.fda.gov/downloads/Drugs/Guidance Compliance Regulatory Information/Guidances/UCM358301. Pdf.
[3] Fast Track Drug Development Programs – Designation, Development, and Application Review [EB/OL]. (2016-12-20). http://www. fda. gov/Biologics Blood Vaccines/Guidance Compliance Regulatory Information/ProceduresSOPPs/ ucm073515. Htm.
[4] Dolgin E. Cancers true breakthroughs[J]. Nat Med, 2013, 19(6): 660-663.
[5] Food and Drug Administration Safety and Innovation Act[EB/OL]. (2016-12-20). https://www. gpo. gov/fdsys/pkg/ BILLS-112s3187enr/pdf/BILLS-112s3187enr. pdf.
[6] Walker G, 史志祥, 陳菁. 美国新增加速审批通道——突破性药物认定[J]. 药学进展, 2013, 37(11): 605-606.
[7] 董江萍, 李茂忠, 姚立新, 等. 美国FDA用于严重病症的药品和生物制品加快审评计划[J]. 中国新药杂志, 2014, 23(2): 171-183, 202.
[8] CDER Approval Times for Priority and Standard NDAs and BLAs Calendar Years 1993–2016 [EB/OL].(2016-12-31).https://www.fda.gov/downloads/Drugs/Development Approval Process/How Drugsare Developedand Approved/Drugand Biologic Approval Reports/ NDAandBLAApprovalReports/UCM540942.pdf.
[9] FDA Fast Track Program [EB/OL]. (2016-10-15). http:// smart-therapeutics. com/Drug-Pipeline/Rare- NeglectedDiseases/FDA-Fast-Track-Program.
[10] CDER Breakthrough Therapy Designation Requests Received by Fiscal Year [EB/OL]. (2016-02-15). http://www. fda. gov/ downloads/Drugs/DevelopmentApprovalProcess/How Drugs are Developedand Approved/Drugand Biologic Approval Reports/IND Activity Reports/UCM481540.Pdf.
[11] CDER New Drug Review: 2016 Update [EB/OL]. (2016-12-14). https://www.fda.gov/downloads/About FDA/Centers Offices/Office of Medical Products and Tobacco/CDER/ UCM533192.pdf.
[12] CDER Fast Track Products Approved Since 1998 through June 1, 2010 [EB/OL]. (2010-06-01). https://www.fda.gov/ downloads/Drugs/Development Approval Process/How Drugs are Developed and Approved/Drug and Biologic Approval Reports/UCM216527.pdf.
[13] Yang G, Wang Y, Zeng Y, et al. Rapid health transition in China, 1990–2010: findings from the Global Burden of Disease Study 2010[J]. Lancet, 2013, 381(9882): 1987-2015.
[14] 袁林, 邵明立. 美国FDA加快新药审评策略以及对我国的启示[J]. 中国新药杂志, 2015, 24(21): 2401-2404, 2409.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 04:28:00
原子与分子物理学报(2021年1期)2021-03-29 07:28:24
东坡赤壁诗词(2020年2期)2020-06-04 15:44:10
人间(2016年28期)2016-11-10 11:51:06
美与时代·美术学刊(2016年8期)2016-11-09 02:37:40
新闻前哨(2016年10期)2016-10-31 17:28:25
商场现代化(2016年22期)2016-10-18 20:13:24
商场现代化(2016年22期)2016-10-18 20:06:05
今传媒(2016年9期)2016-10-15 22:27:04
中国交通信息化(2016年8期)2016-06-06 03:56:29
