
微波辅助、磷酸催化合成半乳聚糖的结构分析
2017-03-21 10:19王海松丁寅翼成向荣乐国伟
食品与生物技术学报 2017年1期
纪 高, 王海松, 丁寅翼, 成向荣*,2, 乐国伟,2
微波辅助、磷酸催化合成半乳聚糖的结构分析
纪 高1, 王海松1, 丁寅翼1, 成向荣*1,2, 乐国伟1,2
(1.江南大学 食品学院,江苏 无锡 214122;2.食品科学与技术国家重点实验室,江南大学,江苏 无锡214122)
以磷酸催化、微波辅助合成半乳聚糖为研究对象,探讨半乳聚糖的化学结构及聚合反应的区域选择性和立体选择性。综合采用高效凝胶渗透色谱、红外光谱、甲基化分析及一维、二维核磁共振波谱(1H NMR,13C NMR,1H-1H COSY,TOCSY,HSQC,HMBC),对半乳聚糖的相对分子质量、糖苷键的立体构型和连接位点进行表征。结果表明,反应合成的半乳聚糖为多分支结构,重均相对分子质量为2 657,重均聚合度为16,糖残基以α-型半乳吡喃糖为主,主要为→4)-α-D-Galp(1→和→2,4)-α-D-Galp(1→片段。结构分析结果进一步表明,微波辅助、磷酸催化半乳糖缩合过程中,不同位点的羟基具有不同的反应活性,4位羟基的反应活性最强。
微波;磷酸;半乳聚糖;结构分析
半乳聚糖是一类广泛存在于海洋藻类、陆生植 物或动物组织中,由半乳糖或以半乳糖为主、其它单糖为辅构成的直链或支链聚合物[1]。半乳聚糖,尤其是来源于藻类的半乳聚糖硫酸酯,具有增强免疫、抗病毒、抗凝血及抑制肿瘤转移的生物学功能[2-5]。此外,半乳聚糖作为一类膳食纤维,还可以有效促进肠道乳酸菌增殖,提高肠道短链脂肪酸的含量[6]。
目前,半乳聚糖的制备主要依赖于天然资源的提取、水解和衍生。天然资源的生长周期性和提取工艺的复杂性严重制约了半乳聚糖的深入开发和应用。结合糖在酸催化、加热条件下可脱水缩合的特点,以及微波辐射具有加热均匀、快速的优势,作者研究了微波辅助快速合成低聚糖、多糖的技术,并分别以葡萄糖、甘露糖、半乳糖为原料,在杂多酸催化下,快速、高收率地合成了相应低聚糖或多糖[7-9]。糖类物质的化学结构中普遍存在多个位点的异构,包括区域异构及立体异构,即糖苷键不同连接位置、不同立体化学的异构形式,是化学法合成低聚糖、多糖过程中面临的主要挑战[10-12],也是微波辅助合成聚糖过程中亟待解决的科学问题。若能够掌握微波辐照、酸催化单糖聚合过程中的区域选择性和立体选择性规律,那么,微波辅助定向合成低聚糖、多糖将有可能成为现实。
王海松等对微波辐照、杂多酸催化半乳糖聚合的反应进行了优化,并对反应产物进行了初步的化学表征,结果发现合成的半乳聚糖平均聚合度为17,半乳糖残基以β-构型为主[9],但糖苷键的连接位置尚未阐明。作者在上述优化条件的基础上,以半乳糖为原料,采用食品中常用的酸化剂——磷酸为催化剂,微波辐照合成半乳聚糖,并对合成产物进行细致的结构分析,总结半乳糖聚合过程中的区域选择性和立体选择性。
1 材料与方法
1.1 材料与试剂
D-半乳糖(Gal)、氯化钠、磷酸、三氟乙酸、无水乙醇(99%):国药集团化学试剂有限公司产品;重水(D2O):上海晶纯生化科技股份有限公司产品;Sephadex G-25葡聚糖凝胶:北京索莱宝科技有限公司产品。
1.2 仪器与设备
XH-200A型电脑微波固液相合成/萃取工作站:北京祥鹄科技发展有限公司产品;BSZ-100型自动部分收集器:上海青浦沪西仪器厂产品;R-205型旋转蒸发仪:无锡申科公司产品;1260型高效液相色谱仪 (配Shodex RI-101示差检测器):美国Agilent公司产品;ICS-5000型离子色谱仪器:美国Dionex公司产品;600型高效凝胶渗透色谱仪 (配2410视差检测器):美国Waters公司产品;7890A-5975C型气相色谱质谱联用仪:美国安捷伦公司产品;560傅里叶变换红外光谱仪:美国Nicolet公司产品;DRX-500核磁共振仪:德国Bruker公司产品。
1.3 方法
1.3.1 半乳聚糖的合成和纯化 半乳聚糖的合成和纯化参考王海松等的方法[9]。25 g D-半乳糖与1.1%磷酸、3.75 mL氯化钠溶液(0.25 mol/L)充分搅拌混匀后送入微波反应器。设定微波功率800 W、反应温度130℃、反应时间4.5 min,开启搅拌器120 r/min,并开启微波合成仪。反应结束后,得淡黄色胶状固体。反应产物溶于去离子水中,经5倍体积的无水乙醇沉淀后,弃上清液。多糖沉淀加去离子水复溶,采用Sephadex G-25葡聚糖凝胶色谱柱分离,以去离子水作洗脱剂,流量0.5 mL/min,用部分收集器收集洗脱液,每管收集量为3 mL,时间间隔6 min。收集液中的糖组分采用硫酸-苯酚法测定[13],检测波长为490 nm,以吸光度为纵坐标,绘制洗脱曲线。收集各管洗脱液,旋转蒸发浓缩后,冻干得白色粉末状样品。
1.3.2 相对分子质量测定 采用高效凝胶渗透色谱法测定半乳聚糖的相对分子质量。标准品和样品溶液:标准葡聚糖Dextran(相对分子质量为828,2 700,9 750,36 800,13 5350,Sigma-Aldrich公司产品);葡萄糖:Sigma-Aldrich公司产品;用流动相配成质量浓度为5 g/L的溶液,然后于5 000 r/min速率下离心10 min,取上清;色谱柱:TSKgel G3000 PWXL凝胶色谱柱(300 mm×7.8 mm,日本TOSOH公司);流动相:0.1 mol/L NaNO2含0.5 g/L NaN3;流量:0.9 mL/min;柱温:45℃;进样量:20 μL。将系列标准葡聚糖和葡萄糖相对分子质量的对数Lg Mw对其相应的保留时间tR进行回归处理,得出线性回归方程。
1.3.3 半乳聚糖红外光谱分析 称取半乳聚糖样品1.3 mg,加入200目的KBr粉末150 mg,于红外灯照射下,在玛瑙乳钵中研磨均匀,装入压片磨具,用油压机以20 MPa的压力压制2 min,然后用镊子小心取下压片,装入样品架,用Nicolet 560傅里叶变换红外光谱仪于500~4 000 cm-1范围内扫描红外光谱。
1.3.4 甲基化分析 半乳聚糖的甲基化分析参考Hakomori的方法[14]。20 mg干燥的半乳聚糖样品溶解于6 mL DMSO中,室温振荡2 h。在氩气保护下加入甲基亚磺酰负离子(30 mg的NaH固体溶解于3 mL无水DMSO中,温度控制在50~55℃,搅拌至反应液呈墨绿色,得甲基亚磺酰负离子),常温下磁力搅拌至颜色退去,冰浴后缓慢加入4 mL碘甲烷,至溶液逐渐变为淡黄色。反应结束后加入少量蒸馏水终止反应,然后将生成物透析,冷冻干燥得甲基化半乳聚糖。向上述甲基化半乳聚糖中加入1 mL甲酸,110℃水解6 h,加热除去甲酸,再加入2 mL浓度为2 mol/L的三氟乙酸,置于安瓿瓶中,充惰性气体密封后100℃反应6 h,氩气流中冷却、干燥。在水解产物中加0.6 mL去离子水和50 mg NaBH4,室温下过夜,加少量甲酸至无气泡,加甲醇蒸干。然后加入1 mL的吡啶-乙酸酐(体积比为1∶1),100℃反应2 h,旋蒸干燥后加氯仿溶解,进行GC-MS分析,进样量为1~2 μL。GC-MS分析色谱柱:Agilent DB-5ms(30 m×0.25 mm×0.25 μm);载气流量(氦气>99.99%):1.0 mL/min,分流比1∶50;程序升温:80℃,保留5 min,以15℃/min升至200℃保留l min,再以10℃/min升至260℃,保留10 min;接口温度250℃;EI+源:70 eV,250℃,扫描频率5次/秒,质量范围:33~600 amu。数据分析采用AMSD化学工作站软件(version E.02.02.1431)和NIST11数据库。
1.3.5 核磁共振波谱分析 半乳聚糖样品前处理参考Hu等的方法[15]。25 mg半乳聚糖溶解于0.4 mL重水中,振荡15 min,溶解后冻干,该步骤重复操作3次。最后将冻干的半乳聚糖样品溶解在0.4 mL重水中,Bruker DRX-500核磁共振光谱仪检测,记录1H-NMR,13C-NMR,1H-1H COSY,TOCSY,HMBC,HSQC谱。1H-NMR和13C-NMR光谱测定频率分别为500.13和125.77 MHz,样品测定温度65℃。
2 结果与讨论
2.1 半乳聚糖的制备
中强酸可以促进糖苷键原子的质子化,加快多糖的水解和单糖的缩合。王海松等[9]采用微波辅助、杂多酸催化方法快速合成了半乳聚糖,并对合成工艺进行了优化。在本实验中,根据优化后的反应条件,采用食品工业中常用的磷酸代替杂多酸来催化半乳糖的聚合。反应产物经过除杂、柱色谱分离,得到半乳聚糖22.6 g,反应得率为90.4%,与文献[9]结果相近。实验结果表明,磷酸能够快速催化半乳糖聚合,与杂多酸相比,磷酸具有引入杂原子更少、反应产物更易处理的优势。
2.2 相对分子质量测定
根据系列标准葡聚糖、葡萄糖溶液的保留时间,以保留时间tR对相对分子质量的对数lg Mw作图,结果见图1。多糖相对分子质量在180~135 350范围内,Lg Mw与tR呈良好线性关系,回归方程为Lg Mw=0.437 7tR+12.368,R2=0.998 8。
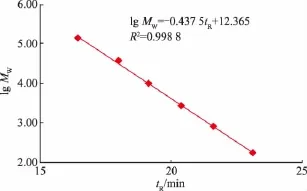
图1 葡萄糖和葡聚糖标准曲线Fig.1 Calibration curve of glucose and dextrans
经过乙醇沉淀和柱色谱分离纯化后,半乳聚糖的高效凝胶渗透色谱行为如图2所示,为单一、狭窄、对称的色谱峰,表明制备得到的半乳聚糖纯度较高。分析结果表明,半乳聚糖的重均相对分子质量(Mw)为2 657,数均相对分子质量(Mn)为2 215,峰位相对分子质量(Mp)为2 196,重均聚合度为16,分子质量分布系数(HI=Mw/Mn)为1.19。HI值趋近于1,表明分离纯化后的半乳聚糖分子质量的多分散程度小,合成制备的半乳聚糖分子质量分布较为均一。
2.3 红外光谱分析
实验合成的半乳聚糖和D-半乳糖的红外光谱图如图3所示。与D-半乳糖相比,半乳聚糖位于3 000~3 700 cm-1处的O—H伸缩振动吸收峰较为宽钝,提示半乳聚糖中存在丰富的、不同化学环境的羟基基团。王海松等认为1 647 cm-1处是C=O振动吸收峰[9],但C=O振动一般出现在1 725~1 730 cm-1处,这与红外光谱信号并不一致,并且在1H和13C NMR图谱(图4)中未观察到与羰基或羧基相对应的化学位移信号。水的特征吸收峰出现在3 420 cm-1和1 630 cm-1附近,故而推测1 647 cm-1处是水的吸收峰,表明半乳聚糖中存在微量的水分。D-半乳糖和半乳聚糖在1 000~1 200 cm-1处存在3个强吸收峰,表明两者均为吡喃型糖苷。877 cm-1和793 cm-1处的吸收峰表明半乳聚糖中存在α-和β-构型的半乳糖残基[9]。

图2 半乳聚糖的高效凝胶渗透色谱分析Fig.2 High performance gel permeation chromatography of galactan
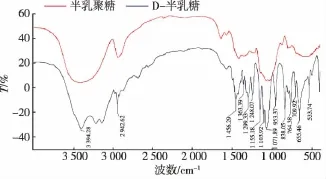
图3 微波辅助合成半乳聚糖和D-半乳糖的红外光谱图Fig.3 FT-IR spectra of synthesized galactan and galactose
2.4 甲基化分析
从半乳聚糖的甲基化结果(表1)可以看出,该多糖中半乳糖的主要连接方式为1,4和1,2,4连接,比例约为1.5∶1,而文献报道红藻来源的半乳聚糖的主链以1,3和1,4连接为主[16-17]。此外,微波辅助合成的半乳聚糖结构中还存在部分的1,6连接型半乳糖残基,以及少量的1,3和1,4,6连接的半乳糖结构片段,而1,2和1,3,6连接的半乳糖残基含量极低。这些结果表明,在反应过程中,半乳糖上不同位置羟基的反应活性不同,4-OH>6-OH>3-OH>2-OH。2,3,4,6-Me4-Gal为半乳糖末端,含量为4.2%,表明半乳聚糖具有较高的聚合度,与相对分子质量的测定结果一致。
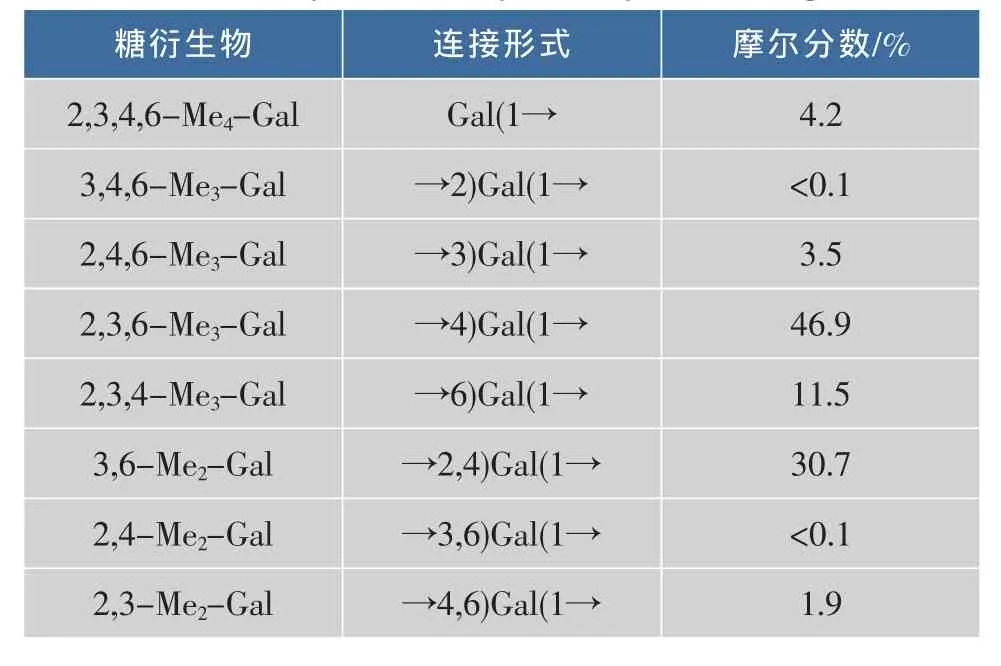
表1 半乳聚糖的甲基化分析Table 1 Methylation analysis of synthesized galactan
2.5 核磁共振波谱分析
核磁共振波谱是多糖结构解析过程中的一个强有力的工具,能够提供更多糖残基的平面结构和立体构型信息。在半乳聚糖的1H-NMR谱(图4(a))中,大多数的端基质子化学位移出现在δ 5.02~5.30(J=0~2 Hz)处,而少数端基质子信号位于δ 4.49(d,J=5.5 Hz)和δ 4.63(d,J=6.0 Hz),δ 3.5~4.4为半乳糖残基H-2~H-6的质子信号。一般地,在以重水为溶剂的核磁测试中,吡喃型糖残基的α-构型端基质子出现在δ 5~6,偶合常数为2~4 Hz;而β-构型端基质子出现在δ 4~5,偶合常数为6~8 Hz[18]。因此,合成半乳聚糖主要以α-构型为主,同时有少量的β-构型半乳糖残基。这与王海松等[9]的分析结果截然相反,可能是由于不同氘代试剂对质子化学位移的影响,从而导致分析结果不一致。在王海松等合成的半乳聚糖的1H NMR图谱[9]分析中,采用氘代二甲基亚砜为溶剂,致使端基质子的核磁信号集体往高场移动约0.3×10-6,却以重水为测试试剂时的构型判别规律为参照,这是不恰当的。在13CNMR谱中,半乳聚糖的端基碳化学位移主要集中在δ 99~110,而C-2~C-6的信号集中在δ63~85,未观察到羰基或羧基的化学位移信号(图4(b))。
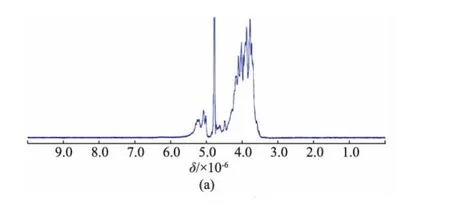
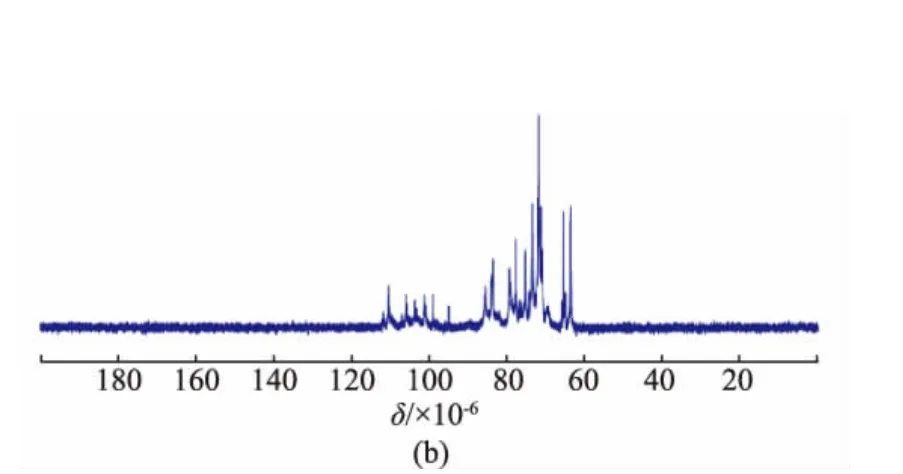
图4 聚半乳糖的1H(500 MHz)和13C(125 MHz)核磁共振谱图Fig.41H and13C NMR spectra of synthesized galactan(500 MHz for1H NMR and 125 MHz for13C NMR in D2O)

图5 半乳聚糖的1H-1H COSY和TOCSY核磁共振谱图Fig.51H-1H COSY and TOCSY spectra of synthesized galactan

图6 半乳聚糖的HSQC和HMBC核磁共振谱图Fig.6 HSQC and HMBC spectra of synthesized galactan
根据半乳糖残基H-1/H-2/H-3/H-4/H-5/H2-6质子的相关性,分析反映邻近质子偶合相关性的1H-1H COSY谱和反映同一自旋体系中质子标量偶合相关性的TOCSY谱(图5),对不同连接型半乳糖残基的质子信号进行归属,结果如表2所示。以β-构型半乳糖残基端基质子信号δ 4.49为例,在TOCSY谱中,δ 4.49与δ 3.60,3.70,3.98,4.02有较强的相关信号,表明这5个质子存在于同一个半乳糖残基中;结合1H-1H COSY谱中δ 4.49与3.60的相关信号,推断H-2的化学位移为δ 3.60,进而根据H-2/H-3的相关信号,推断H-3的化学位移为δ 3.70;类似地,最终推导了整个半乳糖残基的质子信号。根据HSQC谱中氢、碳核磁信号的相关性(图6(a)),归属了不同质子信号对应的碳化学位移(表2),并根据HMBC谱中(图6(b)),氢、碳信号之间的长程偶合(3J)相关,对归属的质子信号进行验证,并推断半乳糖残基中的取代位点。端基质子δ 5.02与δ 73.5(C-6)有强HMBC相关信号,表明半乳聚糖中存在6位羟基取代的糖残基,而往低场移动了约8×10-6的C-6铘化学位移,进一步表明结构中存在→6)Gal(1→结构片段。
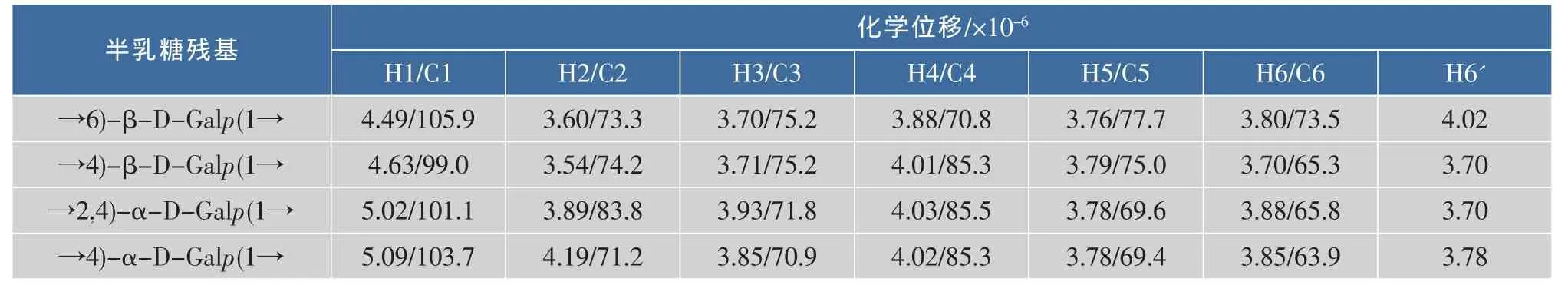
表2 半乳聚糖主要糖残基的化学位移数据Table 2 Chemical shifts of major galactosyl residues in synthesized galactan
对一维 (1H,13C NMR)和二维 (1H-1H COSY,TOCSY,HSQC和HMBC)核磁图谱的综合分析,最终解析了→6)-β-D-Galp(1→,→4)-β-D-Galp(1→,→4)-α-D-Galp(1→和→2,4)-α-D-Galp(1→糖残基信号(表2),对照半乳聚糖的相对分子质量和甲基化分析结果(表1),表明微波辅助、磷酸催化合成的半乳聚糖结构中存在较多的→4)-DGalp(1→和→2,4)-α-D-Galp(1→片段,推测该糖链为多分支的链状结构。
3 结语
在微波辐照条件下,采用磷酸代替杂多酸能够快速、高效地催化D-半乳糖的缩合,纯化后半乳聚糖的得率为90.4%。反应生成的半乳聚糖为分支链状结构,平均聚合度为16,立体构型以α为主,新糖苷键的生成主要发生在4位、2位和6位的羟基上。通过对半乳聚糖的结构分析表明,微波辅助、磷酸催化半乳糖缩合过程中,不同位置的羟基反应活性不同,若能对目标羟基进行保护性修饰,则有可能进一步提高反应的选择性。微波辅助合成半乳聚糖结构的阐明,也为进一步研究和理解其生物活性提供了基础。
[1]CEDRIC D,TARATRA Andree F,PHILIPPE M.Galactans:an overview of their most important sourcing and applications as natural polysaccharides[J].Brazilian Archives of Biology and Technology,2011,54(6):1075-1092.
[2]CARLUCCI M J,SCOLARO L A,ERREA M I,et al.Antiviral activity of natural sulphated galactans on herpes virus multiplication in cell culture[J].Planta Medicine,1997,63(5):429-432.
[3]CHATTOPADHYAY K,MATEU Cecilia G,MANDAL P,et al.Galactan sulfate of Grateloupia indica:isolation,structural features and antiviral activity[J].Phytochemistry,2007,68(10):1428-1435.
[4]FARIAS W R,VALENTE A P,PEREIRA M S,et al.Structure and anticoagulant activity of sulphated galactans:isolation of a unique sulphated galactan from the red algae Botryocladia occidentalis and comparison of its anticoagulant action with that of sulphated galactans from invertebrates[J].Journal of Bological Chemistry,2000,275(38):299-307.
[5]COOMBE D R,PARISH C R,RAMSHAW I A,et al.Analysis of the inhibition of tumour metastasis by sulphated polysaccharides[J].International Journal of Cancer,1987,39(1):82-88.
[6]WU Chengfei,LI Yan,LE Guowei,et al.Effect of the composing and monosaccharides ratio changing of oligosaccharides on free radical scavenging activity and Lactobacillus proliferation[J].Science and Technology of Food Industry,2014,35:49-56.(in Chinese)
[7]WANG H,SHI Y,LE G.Rapid microwave-assisted synthesis of polydextrose and identification of structure and function[J]. Carbohydrate Polymers,2014,113:225-230.
[8]WANG H,CHENG X,SHI Y,et al.Preparation and structural characterization of poly-mannose synthesized by phosphoric acid catalyzation under microwave irradiation[J].Carbohydrate Polymers,2015,121:355-361.
[9]WANG Haisong,SHI Yonghui,LE Guowei.Microwave-assisted synthesis and structural analysis of galactan[J].Food Science,2014,35:35-39.(in Chinese)
[10]WONG C H.Protein glycosylation:new challenges and opportunities[J].Journal of Organic Chemistry,2005,70:4219-4225.
[11]HSU C H,HUNG S C,WU C Y,et al.Toward automated oligosaccharide synthesis[J].Angewandte Chemie,International Edition,2011,50:11872-11923.
[12]CHENG Xiangrong,WANG Wei,SHI Yonghui,et al.Microwave assisted synthesis of mannose oligosaccharide intermediates[J]. Journal of Food Science and Biotechnology,2015,34(1):21-27.(in Chinese)
[13]GUO Jinlong,CHEN Youjun,SUN Guoqin,et al.Study on phenol-sulfuric acid method for determination of polysaccharide content in Pleurotus eryngii[J].Food Science,2008,29:555-558.(in Chinese)
[14]HAKOMORI S.Rapid permethylation of glycolipids and polysaccharides,catalyzed by methylsulfinyl carbanion in dimethyl sulfoxide[J].Journal of Biochemistry,1964,55(2):205-208.
[15]HU P,XUE R,LI Z,et al.Structural investigation and immunological activity of a heteropolysaccharide from Sargassum fusiforme [J].Carbohydrate Research,2014,390:28-32.
[16]ZHANG Q,LI N,LIU X,et al.The structure of a sulfated galactan from Porphyra haitanensis and its in vivo antioxidant activity [J].Carbohydrate Research,2004,339:105-111.
[17]PEREIRA M G,BENEVIDES N M B,MELO M R S,et al.Structure and anticoagulant activity of a sulfated galactan from the red alga,Gelidium crinale.Is there a specific structural requirement for the anticoagulant action?[J].Carbohydrate Research,2005,340:2015-2023.
[18]CUI S W.Food carbohydrates chemistry,physical properties,and applications[M].CRC Press:Taylor&Francis Group,2005:154-155.
会议消息
Preparation and Structural Characterization of Galactans Synthesized by Phosphoric Acid Catalyzation under Microwave Irradiation
JI Gao1, WANG Haisong1, DING Yinyi1, CHENG Xiangrong*1,2, LE Guowei1,2
(1.School of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi 214122,China)
Galactans synthesized by phosphoric acid catalysis and microwave irradiation were studied for their chemical structures and regio-and stereo-selectivities during the polymerization.A series of analyses including high-performance gel-permeation chromatography,infrared spectroscopy,methylation analysis and NMR spectroscopies(1H,13C,1H-1H COSY,TOCSY,HSQC,HMBC)were used.Results showed that the degree of galactosyl polymerization was 16 and the -galactopyranosyl residues were dominant mainly with→4)-α-D-Galp(1→and→2,4)-α-D-Galp(1→fragments.Structural analyses further indicated that the activities of hydroxyls in galactose were different and the 4-OH exhibited the highest potency.
microwave,phosphoric acid,galactan,structural analyses
TS 245.9
A
1673—1689(2017)01—0027—07
2015-03-24
国家“十二五”科技支撑计划项目(2012BAD33B05)。
*通信作者:成向荣(1985—),男,浙江永康人,医学博士,副教授,主要从事食品营养与功能因子研究。E-mail:cheng-xiangrong@hotmail.com
纪 高,王海松,丁寅翼,等.微波辅助、磷酸催化合成半乳聚糖的结构分析[J].食品与生物技术学报,2017,36(01):27-33.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
宝藏(2021年4期)2021-05-27
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中成药(2018年8期)2018-08-29
中成药(2018年3期)2018-05-07
中华老年多器官疾病杂志(2016年7期)2016-04-28
池州学院学报(2015年3期)2016-01-05
中国塑料(2015年5期)2015-10-14
天津科技大学学报(2015年2期)2015-08-09
国际心血管病杂志(2015年5期)2015-02-27
