
表现为帕金森综合征的肌张力障碍临床研究(附1例利用二代测序方法筛查报告)
2017-03-08 09:35:46万志荣商梦晴冯涛
临床神经病学杂志 2017年1期
万志荣,商梦晴,冯涛
·学术交流·
表现为帕金森综合征的肌张力障碍临床研究(附1例利用二代测序方法筛查报告)
万志荣,商梦晴,冯涛
目的 探讨表现为帕金森综合征(PKS)的肌张力障碍的临床特点。方法 回顾性分析1例表现为PKS的肌张力障碍患者的临床资料。结果 患者为青年女性,31岁起病,临床表现为右足跛行、震颤,不对称起病,应用左旋多巴短期有效,多巴胺转运体(DAT)PET双侧豆状核后部DAT分布明显减少,考虑不典型帕金森病,较长时间按PKS治疗。后利用二代测序法发现DYT6基因发生c.268-4T>A (NM_018105)杂合突变,突变方式为剪切突变,并经一代测序验证。结论 肌张力障碍患者有时临床表现类似PKS,尤其需要与青年型帕金森病合并局灶性肌张力障碍相鉴别。临床观察、基因检测是鉴别的重要方式。
帕金森综合征; 肌张力障碍; DYT6型;二代测序; 一代测序
近年来,越来越多的临床研究[1-4]报道表明:40岁以前发病的青年型帕金森病(YOPD)约14%~50%可发生肌张力障碍,且以足部肌张力障碍表现较常见。马敬红等[3]指出女性、起病年龄越早、统一PD评分量表(UPDRS)总分分值越高PD越易出现足肌张力障碍,且性别与足的肌张力障碍的相关性最高。Jankovic等[4]对其运动障碍诊疗中心200例临床诊断PD患者分析发现,有38%患者首诊时表现出肌张力障碍,且多数PD诊断是在肌张力障碍发生的10年内。使得临床上对于YOPD诊断范围容易扩大,造成误诊。经典PD在英国脑库PD诊断标准中,无论支持还是排除诊断标准中均无肌张力障碍表现。现通过报道1例应用一、二代测序平台筛查的DYT6型肌张力障碍患者的临床资料进行分析,以探讨肌张力障碍与表现为肌张力障碍的YOPD的异同点。
1 临床资料
1.1 一般资料 患者,女,32岁,因“右脚跛行1年余,加重伴头颈、上躯干不自主运动1月”于2014年12月20日就诊。患者1年余前(2013年10月)出现右脚跛行(足内翻),伴右手刷牙欠灵活,行足部MRI、头颅MRI、腰椎MRI无异常。当地骨科考虑骨代谢紊乱,肌注降钙素,口服葡立胶囊等无效,右足跛行逐渐加重。当年12月出现右手抖动,旋转时出现,安静时不明显。后就诊神经内科,行EMG震颤分析右手震颤节律9 Hz;多巴胺转运体(DAT)PET示双侧尾状核尾部、双侧豆状核后部DAT分布明显减少,诊断考虑:YOPD。 服用息宁,每次1片,3次/d,右手灵活性改善,但右足跛行无缓解,且服用息宁1周后出现头颈、上躯干不自主晃动(平躺、侧卧最明显)。就诊于北京某三甲医院,仍考虑YOPD,加用咪多比,患者右脚跛行明显好转,行走接近正常,身体晃动未缓解,但药效仅维持20 d后再次出现右足跛行,且合并后背部疼痛感。后就诊于四川某三甲医院,考虑帕金森综合征合并全身型肌张力障碍,加用利培酮,患者身体晃动明显减轻,但出现头晕,月经紊乱(提前、量极少),并行肉毒素注射(颈部、背部、右小腿),背部疼痛消失,但跛行加重。后就诊于我院门诊,将利培酮逐渐减量,头晕消失,但身体晃动再次出现。加安坦、巴氯芬后跛行有所改善。进一步诊治收入院。既往史:否认嗅觉下降、便秘等;父亲已故,追问病史生前有类似表现,母亲无神经系统异常表现。查体:K-F环(-),意识清楚,无构音障碍,高级智能无异常,头颈部、上躯干不自主晃动,右足跛行,足内翻,双手摆动可,右手姿势性震颤,轮替动作较对侧欠灵活,显笨拙,四肢肌张力不高,腱反射对称活跃,双下肢病理征阴性,颈无抵抗。辅助检查:血常规、肝肾功能、铜蓝蛋白未见异常;汉密尔顿焦虑量表12分,汉密尔顿抑郁量表8分,MMSE评分30分;DATPET:双侧尾状核尾部、双侧豆状核后部DAT分布明显减少,左侧明显;震颤EMG分析9 Hz的节律性震颤点位改变;右足MRI、头颅MRI未见异常(图1)。为明确诊断,行基因检测。
1.2 二代测序方法筛查
1.2.1 血液标本的采集和DNA提取 取静脉血2 ml,EDTA抗凝,常规酚-氯仿方法提取基因组DNA。
1.2.2 遗传性帕金森基因及肌张力障碍基因检测 (1)基因捕获。遗传性帕金森基因及肌张力障碍基因芯片(Agilent),基因列表(共检测帕金森及肌张力障碍相关基因35种);(2)基因测序。测序前,外显子文库使用Agilent 2100检测文库片段大小,片段大小在350~500 bp;测序仪器为Illumina HiSeq 2500;(3)生物信息学分析。①SNP分析流程:测序仪获取原始短序列;去除测序数据中的接头和低质量数据等;把短序列用SOAPaligner软件定位到人类基因组数据相应的位置上;统计测序结果信息,短序列数量、目标区域覆盖大小、平均测序深度等;SOAPsnp用于在目标区域找出位点的基因型,过滤低质量值(质量值≤20)和低覆盖度(深度≤10)的SNP;利用CCDS、人类基因组数据库、dbSNP信息对SNP进行注释,确定突变位点发生的基因、坐标、mRNA位点、氨基酸改变、SNP功能(错义突变/无义突变/可变剪切位点)、SIFT预测SNP影响蛋白功能预测等(参考http://soap.genomics.org.cn/)②InDel分析流程:把去除接头序列和低质量的测序数据用Burrows-Wheeler Aligner (BWA)比对到人类基因组上,用GATK软件找出序列中所含有的插入/缺失(InDel)的信息(参考http://bio-bwa.sourceforge.net);利用CCDS、人类基因组数据库、dbSNP信息对InDel进行注释,确定突变位点发生的基因、坐标、mRNA位点、编码区域序列的改变、对氨基酸的影响、InDel功能(氨基酸插入/氨基酸缺失/移码突变);③采用数据库:HGMD pro,1000g,ESP6500,Inhouse,np138NonFlagged。
共检测帕金森及肌张力障碍相关基因35种,发现DYT6基因发生c.268-4T>A(NM_018105)杂合突变,突变方式为splicing(剪切突变),并经一代测序验证,HGMDpro数据库未见此突变位点报道,其母c.268-4T>A(NM_018105)无突变。被检样品未检测到PD相关基因拷贝数变化和碱基变异。(见图2)。
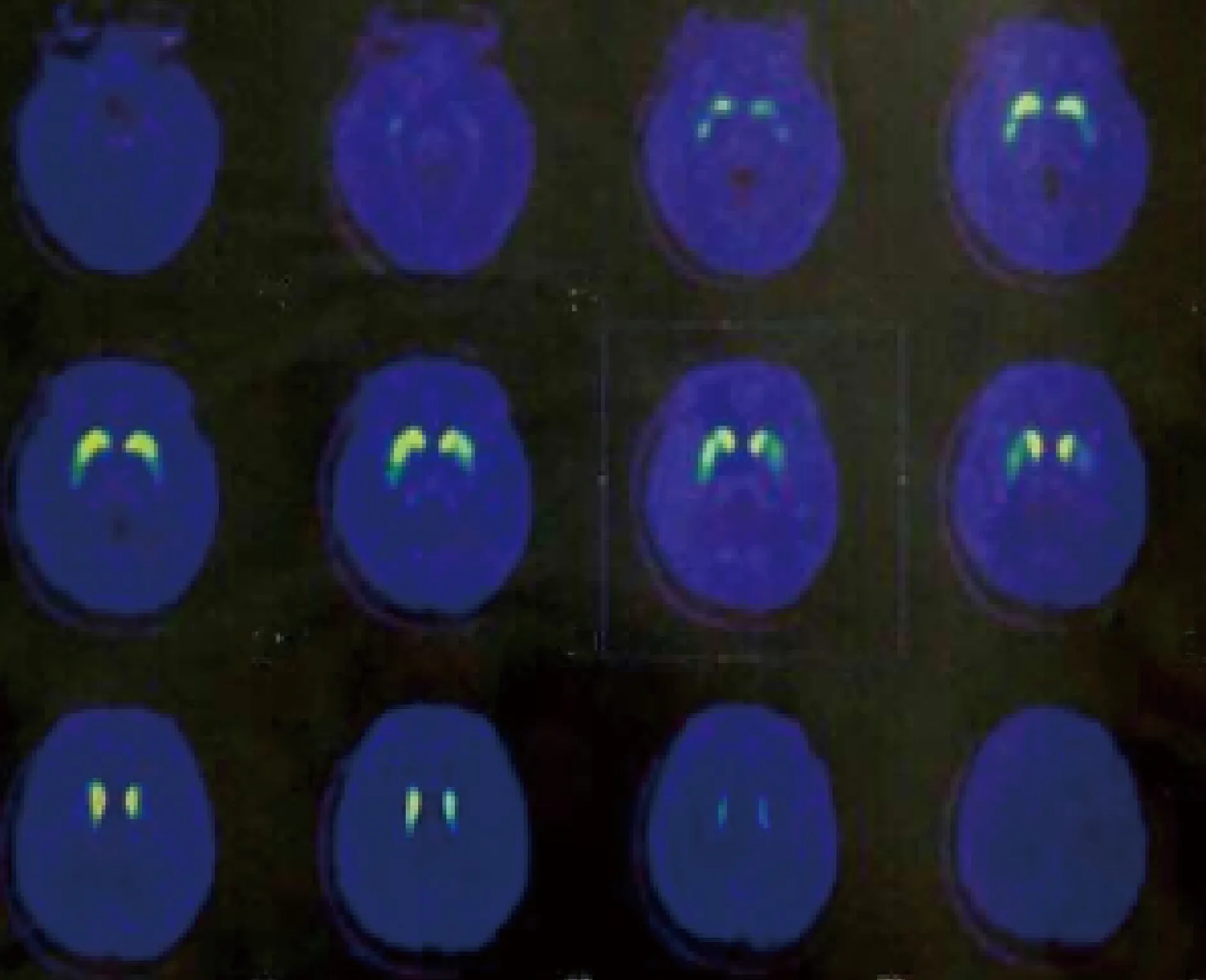
图1 DATPET结果。双侧尾状核尾部、双侧豆状核后部DAT分布明显减少,左侧明显
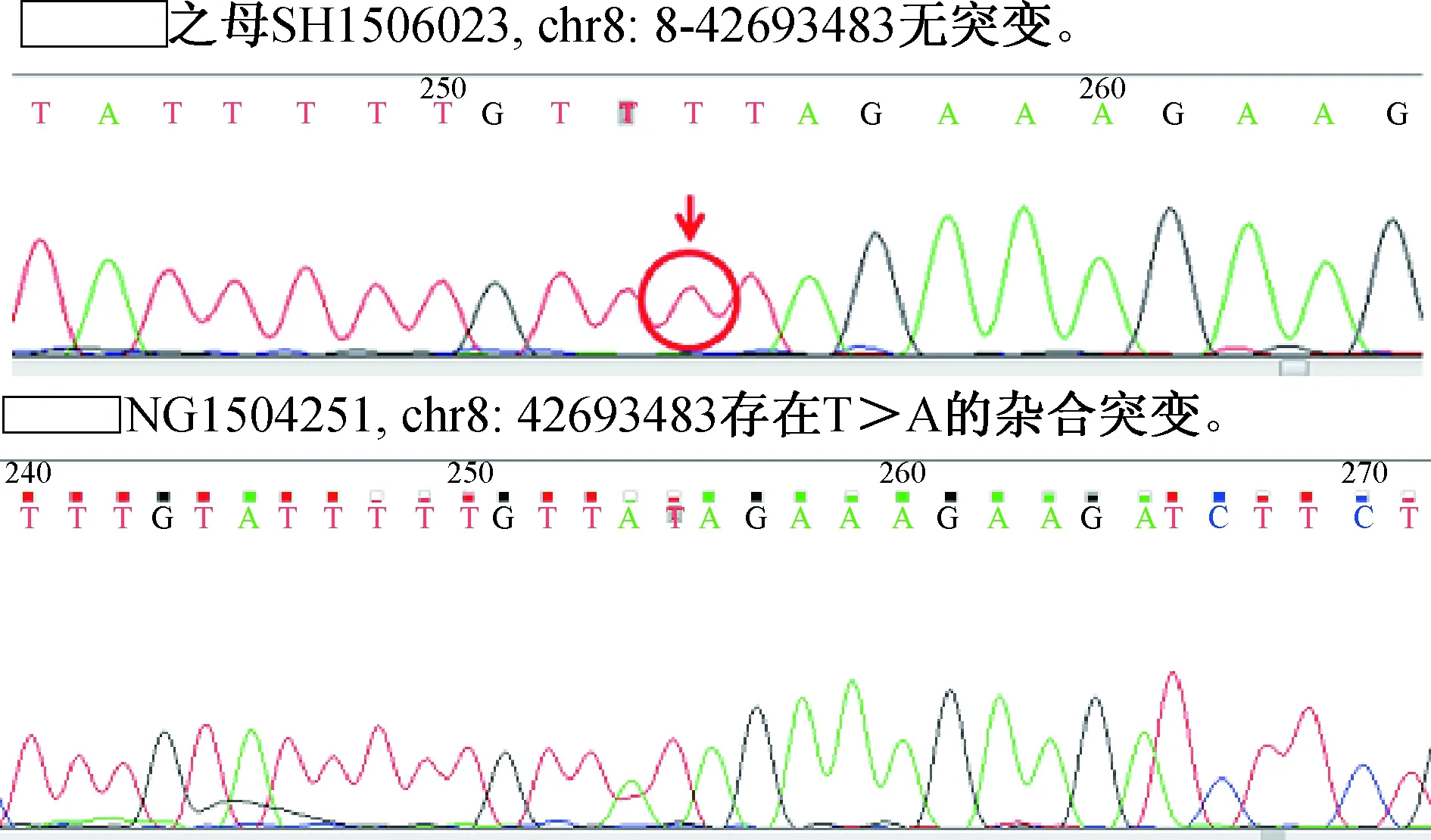
图2 对患者本人及母亲进行一代测序验证,患者chr8-42693483存在突变,其母亲chr8-42693483无突变。
2 讨论
肌张力障碍根据基因位点和临床表型特点分为DYT1~DYT21,多种基因表型得到确定,比如DYT1、5、6、12基因等[5]。1997年,DYT6型肌张力障碍首次在2个Amish-Mennonite家系中被确定,基因位点定位于8号染色体。王琳等[6]报道11例经基因检测证实的DYT6型肌张力障碍以早发型为主,最常见的起病部位是颈部,其次为右上肢,患者主要表现为局灶型、节段型肌张力障碍,其中l例患者出现左下肢肌张力障碍,不伴有足内翻,而晚发型病情长期稳定。与本例晚发全身型DYT6型肌张力障碍有所不同,该患者以足内翻起病,1年左右病变扩展至头颈部、上躯干,表现为全身型,且对左旋多巴反应短暂,病情进展相对快。王琳等[6]报道的DYT6型基因突变患者均为汉族,来自我国的华北、华东和华中地区,本患者来自西南地区(重庆),二代测序基因检测发现发现DYT6基因发生 c.268-4T>A(NM_018105)杂合突变,突变方式为splicing(剪切突变),并进行一代测序进一步验证。对其母亲进行一代测序无此位点突变。
Xiao等[7]报道新的THAP1突变具有较大的临床异质性,他们收集1114名成年发病的原发性肌张力障碍患者,发病年龄在8~69年(平均48年),测序确定了6个新的错义突变,认为THAP1基因发生序列变异是导致他们发生家族性,特别是早发性肌张力障碍的原因。并由此得出结论,THAP1基因上发生的各种各样的序列的变异,与解剖分布(基因、染色体等)以及家族性、散发性的肌张力障碍的发病年龄有关。本例患者父亲已故,追问病史,生前曾有类似临床表现,该患者属于遗传早现。临床上,DYT6型肌张力障碍需重点与以局限性肌张力障碍临床表现起病PD、多巴反应性肌张力障碍(DRD)等疾病相鉴别[8]。DRD是一组由多种酶代谢缺陷所致、临床以肌张力障碍和帕金森综合征为主、症状波动性、多巴类制剂治疗显著有效的遗传性肌张力障碍疾患。根据国际原发性肌张力障碍遗传性分类,DYT5、DYT14均为DRD[8]。对于已治疗PD的肌张力障碍,大多由于病情进展及多巴制剂引起,增加左旋多巴剂量会在频率和程度上加重PD患者肌张力障碍症状,临床容易识别。但对于未经治疗PD,尤其是YOPD以局限性肌张力障碍临床表现起病,有时难于区分[1]。YOPD患者中足部肌张力障碍典型表现为运动后诱发足部从足趾开始的抽筋样不适,甚至足部扭转、跛行,称之为“运动源性肌张力障碍足”。现已明确青年发病的PD是由不同致病基因突变所致遗传性帕金森综合征,主要包括parkin基因(PARK-2)突变所致以常染色体隐性遗传性青少年型帕金森综合征(JP)以及PARK-6、DJ-1(PARK-7)等基因突变所致早发性帕金森综合征[2,9]。LeWitt等[10]曾报告部分患者肌张力障碍在出现帕金森症状之前0.5~20年,累及颈、上肢、口颌、单侧下肢或足部的肌张力障碍(PD症状较重侧)。Tolosa等[2]对PD患者出现肌张力障碍的研究文献总结提出未治疗PD患者可表现有不同类型肌张力障碍,常为局灶性,包括单侧马蹄内翻足样、上肢-前臂或前臂-手屈曲、书写痉挛、口颌肌张力障碍、痉挛性斜颈、或多种类型合并;还可表现出非典型肌张力障碍,包括PD书写痉挛、驼背样躯干前屈、肛门痉挛等。严新翔等[11]指出,YOPD患者具有以下特点:(1)PD三联征(运动迟缓、肌强直、静止性震颤)均较轻,早期症状常不典型;(2)局限性肌张力障碍常见,尤以足部肌张力障碍明显;(3)腱反射活跃或亢进常见;(4)症状波动常见,如晨轻暮重和睡眠后症状可减轻等;(5)病程长,病情进展缓慢;(6)对多巴制剂治疗反应良好,但由多巴制剂引起的运动障碍和症状波动常见。随着近年来[11C]CFT脑DATPET在临床上的运用,其对PD的早期诊断及病情严重度评估提供了很好的依据,但实际上在多系统萎缩、进行性核上性麻痹等帕金森叠加综合征患者中,PET也可以存在纹状体的DAT摄取值下降[12]。冼文彪等[13]提出,需结合[18F]FDG脑代谢,才能进一步提高PD诊断及鉴别率,PD患者在苍白球、丘脑、脑干、小脑、感觉运动皮质区代谢增强和运动前皮质、顶枕区代谢减弱。Eckert等[14]研究发现,这种代谢模式为PD特有的脑功能网络代谢模式,在正常人、帕金森叠加综合征和继发性帕金森综合征患者均不存在。DYT6型也可有DAT摄取值下降,提示存在黑质纹状体通路受损,可能还有需要结合FDGPET、D2受体等检查综合分析,并扩大样本进一步观察DATPET在DYT6型中的特点。本例患者对左旋多巴药物疗效反应一般,服用20 d后出现全身晃动,且与体位有关,考虑并非异动症而是全身型肌张力障碍的表现。因此,临床上疑似YOPD患者如具有以下特点需重新考虑诊断:(1)对多巴胺能药物反应不良或短暂有效;(2)病情进展相对快;(3)肌张力障碍由足部进展为全身,需要重点鉴别感觉诡计或是药物诱导的运动障碍。故仔细的临床查体、询问病史,区分药物的反应性及时效性、随访患者在鉴别上尤为重要,DATPET检测只能是诊断的一种手段,而非必备的条件,可结合基因检查、家族史中有无类似患者等协助诊断,但同时也需要对帕金森相关基因进行分析,以做鉴别。
治疗方面,DYT6型肌张力障碍患者对苯海索、巴氯芬和氯硝西泮治疗反应欠佳[6],肉毒毒素可以改善局部症状,符合该患者特点,也是与YOPD、DRD鉴别的要点之一。国外有应用脑深部电刺激治疗DYT6型肌张力障碍,取苍白球内侧为作用位点,对于颈部和肢体肌张力障碍有一定疗效,但个体差异明显[15]。值得一提的是,患者曾服用利培酮后出现头晕、月经量减少,与利培酮引起体位性低血压及血浆中催乳素浓度的增加,从而发生月经失调相关[16]。
[1]张颖冬.帕金森综合征中的肌张力障碍[J].临床神经病学杂志,2015,28:161.
[2]Tolosa E,Compta Y. Dystonia in Parkinson’s disease[J]. J Neurol,2006, 253(Suppl 7):Ⅶ/7.
[3]马敬红, 胡国华, 邹海强,等.影响帕金森病患者出现足肌张力障碍的相关因素[J].中风与神经疾病杂志, 2008,25:322.
[4]Jankovic J,Tintner R.Dystonia and parkinsonism[J].Parkinsonism Relat Disord,2001,8:109.
[5]Ozelius LJ, Lubarr N,Bressman SB. Milestones in dystonia[J].Mov Disord,2011,26:1104.
[6]王琳, 万新华, 成伏波,等.DYT6型肌张力障碍患者的临床表现和影像学特点[J].中华神经科杂志,2013,46:148.
[7]Xiao J,Zhao Y,Bastian RW,et al.Novel THAP1 sequence variants in primary dystonia[J].Neurology,2010,74:229.
[8]Schneider SA,Bhatia KP. Rare causes of dystonia Parkinsonism[J].Curr Neurol Neurosci Rep,2010,10:431.
[9]Koziorowski D,Hoffman-Zacharska D,Slawek J,et al.Incidence of mutations in the PARK2,PINK1,PARK7 genes in Polish early-onset Parkinson disease patients[J].Neurol Neurochir Pol,2013,47:319.
[10]LeWitt PA,Burns RS,Newman RP. Dystonia in untreated Parkinsonism[J]. Clin Neuropharmacol,1986,9:303.
[11]严新翔,郭纪锋,唐北沙,等.青少年型帕金森病60例临床分析[J].卒中与神经疾病,2005,12:227.
[12]Antonini A,Benti R,de Notaris R,et al. 123I-Ioflupane/SPECT binding to striatal dopamine transporter (DAT) uptake in patients with Parkinson’s disease, multiple system atrophy,and progressive supranuclear palsy[J].Neurol Sci, 2003,24:149.
[13]冼文彪,江璐璐,刘妍梅,等.[18F]FDG脑代谢联合[11C]CFT脑多巴胺转运体PET显像对帕金森病的临床研究[J].中国临床神经科学,2014,22:530.
[14]Eckert T,Eidelberg D.Neuroimaging and therapeutics in movement disorders[J].Neuro Rx,2005,2:361.
[15]Panov F,Tagliati M,Ozelius LJ,et al.Pallidal deep brain stimulation for DYT6 dystonia[J].J Neurol Neurosurg Psychiatry,2012.83:182.
[16]朱建忠, 梁颖茵, 朱荣兰,等.利培酮治疗帕金森病合并精神障碍的临床研究[J].吉林医学,2010,31: 3670.
A clinical study on dystonia manifested as parkinsonism(report of 1 next-generation sequencing attached case)
WANZhi-rong,SHANGMeng-qing,FENGTao.
DepartmentofNeurology,AerospaceCentralHospital,Beijing100049,China
Objective To discuss the clinical features of dystonia manifested as Parkinsonism (PKS).Methods Clinical materials of a patient with dystonia manifested as PKS were analyzed retrospectively. Results The onset age of the young women was 31 years old, who was started asymmetrically with symptoms of claudication and tremor of the right foot. Levodopa had a short-term effect. The results of dopamine transporter (DAT) PET showed that DAT in retrolentiform part were decreased significantly. Atypical Parkinson’s disease was considered and she was treated as PKS long-termly. Subsequently, heterozygous mutation of c.268-4T>A (NM_018105) in DYT6 gene was found through the next-generation sequencing, which was a kind of splicing mutation and confirmed by the first-generation sequencing. Conclusions Patients with dystonia might share similar clinical manifestations with PKS. Particularly, they should be differentiated with young-onset Parkinson’s disease combined with focal dystonia. Clinical observation and genetic testing are important approaches to differentiate them.
parkinsonism;dystonia;DYT6 type;next-generation sequencing;first-generation sequencing
100049 北京,航天中心医院神经内科(万志荣,商梦晴);首都医科大学附属北京天坛医院神经病学中心神经变性病科(冯涛)
冯涛
R742.5
A
1004-1648(2017)01-0050-04
2016-03-18
2016-06-08)
猜你喜欢
中国药学药品知识仓库(2022年6期)2022-04-11 22:24:25
老年医学研究(2021年5期)2022-01-19 01:25:18
中国现代神经疾病杂志(2017年1期)2017-03-29 06:39:34
养生保健指南(2016年2期)2016-11-28 09:37:25
广东药科大学学报(2016年6期)2016-03-10 07:33:32
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
中国继续医学教育(2015年6期)2016-01-07 07:38:37
黑龙江八一农垦大学学报(2015年2期)2015-12-08 09:26:15
放射学实践(2015年2期)2015-02-14 05:38:58
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08 06:17:25
