
成人幕上多发原始神经外胚层肿瘤1例及文献复习
2017-02-14 08:38胡重灵娄四龙毛德强戴勤弼
临床神经外科杂志 2017年1期
胡重灵,娄四龙,毛德强,戴勤弼
·临床研究·
成人幕上多发原始神经外胚层肿瘤1例及文献复习
胡重灵,娄四龙,毛德强,戴勤弼
目的 探讨成人幕上原始神经外胚层肿瘤(PNET)的诊断及治疗,提高早期诊断率,改善疗效及预后。方法 回顾性分析重庆市肿瘤医院2015年3月收治的1例成人幕上多发PNET患者。患者影像学检查示颅内多发占位病变,行颅内病灶切除术,术后根据病理检查结果行放疗。并以“PubMed”及“中国知网”为检索工具复习相关文献。结果 患者肿瘤大部分切除,术后病理检查为中枢神经系统原始外胚层肿瘤,行放疗。患者术后4个月时病情进展,术后9个月死亡。文献检索结果显示,国内外2005~2015年报道成人幕上PNET的文献共48篇。其中PubMed搜索文献26篇,病例报告18篇,回顾性分析8篇;“中国知网”搜索文献22篇,病例报告14篇,回顾性分析8篇。共含病例276例,尚未见到成人幕上多发PNET病例的报道。结论 PNET发病率低,恶性程度极高,复发及转移率高;应早期诊断并及时手术治疗,辅以放、化疗综合治疗可延长患者的寿命。
原始神经外胚层肿瘤;幕上;成人
中枢神经系统原始神经外胚层肿瘤(PNET)是一组临床非常少见的恶性颅内肿瘤,在儿童颅内肿瘤中仅占2.5%~6%,发生于成人的更为少见约占0.5%,无明显性别差异,呈浸润性生长,易沿脑脊液循环播散和转移,具有恶性度高、侵袭性强、误诊率高、预后差等特点。近年对PNET的报道逐渐增多,但成人幕上多发PNET尚未见到详细报道,因此对其认识较少,易误诊、延误治疗。本研究回顾性分析重庆市肿瘤医院2015年3月收治的1例成人幕上多发PNET患者的临床资料,并结合文献复习,对该病的临床、病理表现及诊断、治疗和预后进行探讨。
1 临床资料
患者男性,21岁。以“头痛、头晕1个月余,伴复视、肢体无力半个月”于2015年3月入院。患者1个月前无明显诱因出现头痛,伴头晕、恶心、呕吐,头痛逐渐加重,半个月后出现复视、左侧肢体无力、麻木、行走不稳。入院初步诊断为“颅内多发性占位病变,寄生虫?胶质瘤病?转移瘤?”。头颅MRI检查:幕上多发占位病变(图1),左侧额叶、颞叶及胼胝体区域、左枕叶可见多发异常占位性病灶,T1WI呈等低高混杂信号影,T2WI呈等高混杂信号影,边界较模糊,其中额叶病变T1WI见较大的片块状高信号影,似见液平面;病灶最大者约为5.3 cm×4.5 cm,病灶占位效应明显,中线结构向右侧偏移,左侧脑室及左额叶脑实质受压改变;增强扫描病灶明显不均匀环形及不规则强化改变,脑膜强化明显。患者在全麻下行左额叶占位病变切除术,术中见肿瘤囊性部份内含褐色液体考虑有陈旧性出血,实性部份位于左额叶深面,灰红色,质软,部分呈胶冻状,与正常脑组织分界不清,血供极其丰富;病变深达侧脑室前角、胼质体,显微镜下分块大部份切除病变。术后患者接受放疗治疗,因其自身原因未行化疗治疗。术后定期复诊及随访。
术后病理学检查:胚胎性肿瘤,符合PNET(图2)。免疫组化检查:GFAP局灶(+);Syn局灶(+);CD34血管内皮(+);CD56(++);Vim(+);Ki-67(+)40%;Olig-2少数(+);P53(+)80%;MGMT(+);CD99(+);Bcl-2(+);TOPOⅡ耐药基因蛋白(+)20%;NSE局灶(+);CD57小灶(+);Nestin(+);均阳性。
患者术后1个月复查头颅MRI(图3),并进行头部适形调强放射治疗。患者术后4个月时出现头痛、肢体抽搐,复查头颅MRI示病情进展(图4),给予头部钴60放射治疗。术后8个月时再次出现头痛、肢体抽搐、胡言乱语,复查头颅MRI示病情进展(图5),患者未行化疗及其他治疗。术后9个月死亡。

图1 术前头颅MRI平扫+增强
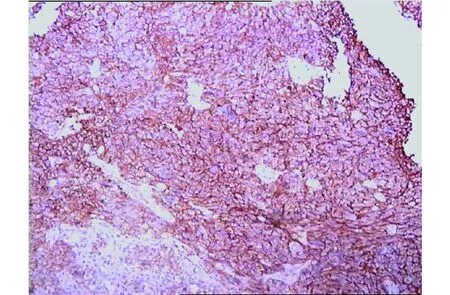
颅内胚胎性肿瘤,符合PNET(中枢神经系统原始神经外胚层肿瘤)。免疫组化:GFAP局灶(+);EMA(-);CK(-);CK低(-);CgA(-);Syn局灶(+);LCA(-);CD34血管内皮(+);CD56(++);Vim(+);Ki-67(+)40%;Olig-2少数(+);Calponin(-);CD163(-);P53(+)80%;S-100(-);MGMT(+);CD99(+);Bcl-2(+);TOPO Ⅱ 耐药基因蛋白(+)20%;SMA(-);Myogenin(-);NSE局灶(+);CD57小灶(+);Fli-l(-);Nestin(+);Desmin(-);TIF1(-);HMB45(-)图2 PNET术后病理学检查、免疫组化(HE染色,×100)

图3 术后1个月头颅MRI平扫+增强
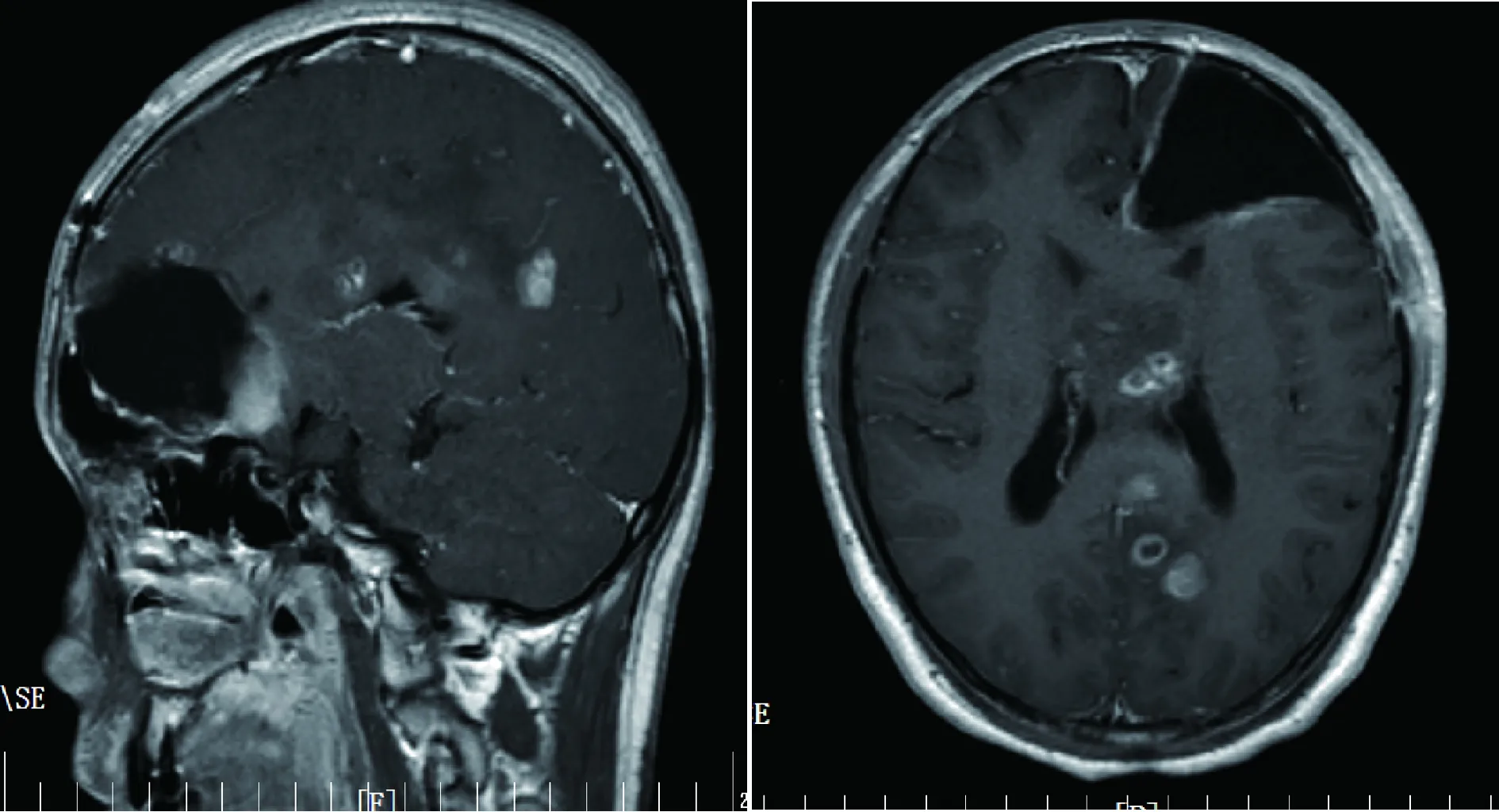
图4 术后4个月头颅MRI平扫+增强

图5 术后8个月头颅MRI平扫+增强
3 讨 论
1973年由Hart和Earle把一组非特异性的小细胞肿瘤定义为PNET,2007年版WHO中枢神经系统肿瘤分类将幕上PNET归类于神经上皮组织的胚胎性肿瘤,包括神经母细胞瘤、神经节细胞神经母细胞瘤、髓上皮瘤和室管膜母细胞瘤。2016版WHO中枢神经系统肿瘤分类[1]是在2007年版本概念和实践上的进一步推进。新的WHO中枢神经系统肿瘤分类首次针对大多数肿瘤在组织学分型基础上增加了分子分型来分类,从而建立了分子时代中枢神经系统肿瘤诊断的新概念。2016年WHO中枢神经系统肿瘤分类主要在弥漫型胶质瘤、髓母细胞瘤和其他胚胎性肿瘤中进行了重分类,这之中包含了结合组织学和分子学特征的新名称定义原则,包括胶质母细胞瘤-IDH野生型和胶质母细胞瘤-IDH突变型;弥漫型中线胶质瘤-H3K27M突变型;RELA融合性室管膜瘤;髓母细胞瘤-WNT激活型和髓母细胞瘤-SHH突变型;胚胎性肿瘤伴有多层细胞菊形团-C19MC激活等。2016版增加了部分新的肿瘤,删除了部分不再与诊断和生物学相关的名称、变化和形式等。其他需要指出的变化还包括在非典型性脑膜瘤中增加脑侵犯作为一个诊断标准,在新的单独神经纤维瘤/血管外皮细胞瘤联合体中引入了软组织类型分级系统,这种分型系统不同于其他中枢神经系统肿瘤分型系统。2016年WHO中枢神经系统肿瘤分类在髓母细胞瘤外的胚胎源性肿瘤分型上做了重大调整,将PNET从诊断词条中删除。许多重新分类基于承认这些少见肿瘤显示19号染色体(19q13.42)C19MC区域的扩增,包括胚胎性肿瘤伴多层菊形团(C19MC变异)、胚胎性肿瘤伴多层菊形团(NOS)、髓上皮瘤(承认一些明显的真正的髓上皮瘤没有C19MC扩增)。目前对于胚胎源性肿瘤的理解正在改变,期望今后分子标记物进一步研究可以使得这些肿瘤及其亚型的诊断更精确。
杨立等[2]研究认为,中枢神经系统PNET是由于具有多向分化潜能的干细胞-原始神经外胚层细胞基因调控失常而出现向神经上皮各个不同阶段的组织分化甚至向间叶组织分化而形成。因此其发病年龄较轻,30岁以下的青少年、儿童多见。肿瘤多位于白质区域,可向脑深部和脑表面生长,甚至破坏颅板,以额叶为最好发部位,其次为顶叶、颞叶、枕叶、基底节区和脑室。本研究以“PubMed”及“CNKI”数据库检索相关文献,检索关键词分别为“audlt supratentorial primitive neuroectodermal tumor”及“成人幕上原始神经外胚层肿瘤”。检索结果显示,2005~2015年PubMed一共收录成人幕上PNET相关文献26篇,其中病例报告18篇,回顾性分析8篇;共193例患者,发病年龄18~88岁,发病时间1~12个月,平均3.9个月,治疗后生存时间48 h~4年,1年生存率61%,3年生存率29%。CNKI收录相关文献22篇,病例报告14篇,回顾性分析8篇,共83例患者;发病年龄18~66岁,发病时间10 d~10个月,平均4.6月,治疗后生存时间1个月~3年,1年生存率64%,3年生存率32%;病死率与国外文献报道的相似。
中枢神经系统PNET的临床表现主要是颅内高压症状及局灶性神经功能障碍,易误诊,与其他颅脑疾病难以鉴别,因此诊断主要依靠病理及免疫组化检查。光镜下90%~95%的肿瘤细胞呈形态不同的未分化状态,呈强嗜苏木素染色,胞浆稀少、核浓染、核浆比高,核分裂象常见,曾有蓝瘤之称。其特征主要包括:光镜下组织细胞属于小圆细胞恶性肿瘤,成巢排列,核周胞浆少,可见有纤维组织分隔或小叶,核染色质颗粒状,核仁不明显,和(或)Homer-Wright菊形团块结构存在及分叶状生长,肿瘤可伴有出血、坏死。免疫组化染色可显示至少1种或2种以上神经分化标志物。细胞遗传学检查可发现染色体异位及融合基因的形成,约40%的中枢神经系统PNET有14q染色体和19q染色体的丢失,另外还有p53基因突变,Neuro D1/D3表达在中枢神经系统PENT常见。Schimidt等[3]提出PNET的诊断标准:至少表达两个不同的神经标志物和(或)有Homer-Wright菊形团结构。其中NSE的表达对诊断PNET具有较高的特异性。免疫组化中MIC-2(+)(即CD99)具有诊断价值[4]。Ki-67阳性也可提高诊断准确率,Kim等回顾分析12例患者中有2/3的患者存在Ki-67阳性,且大体钙化者预后良好,随访未见死亡病例[5]。本例患者存在典型的H-W菊花团,免疫组化显示2种以上神经分化标志物阳性,表明PNET肿瘤细胞具有不同程度的神经元或神经胶质细胞分化倾向。
中枢神经系统PNET为罕见的恶性肿瘤,术前诊断较困难。本例患者术前诊断不清,这与本病罕见及对其认识不足有关。诊断中枢神经系统PNET要将临床症状、影像学检查及病理检查综合起来考虑,病理诊断仍是最终确诊的标准。因中枢神经系统PNET在病理形态上表现为小圆细胞性肿瘤,需从发病年龄、好发部位、病理及免疫组化、影像学特点等几方面与发生在颅内的原发或转移性小圆细胞性肿瘤相鉴别,如髓母细胞瘤、原发性颅内的淋巴瘤、转移性小细胞未分化癌、多形性胶质母细胞瘤等。
目前,中枢神经系统PNET的发病机制尚未完全清楚,但大部分学者认为,这是在神经系统逐步发育成熟的过程中出现了异常分化,多数肿瘤细胞处于原始未分化阶段,形成了PNET。Pang等[6]研究显示,由于启动子的超甲基化,造成DLC-1基因的表达缺失与PNET的形成相关。Inda等[7]研究也显示,RASSF1A基因的突变及其甲基化方式的不同可能是PNET与髓母细胞瘤区别的成因。有研究表明该类肿瘤的发生与染色体移位有关,在PNET患者基因检测过程总发现患者多有染色体移位t(11;22)(q24;q12)[8]。有文献报道颅内肿瘤术后进行常规化疗可诱导继发性PNET的发生[9]。因此可以肯定,PNET的发生、发展是多种因素包括原癌基因的激活、抑癌基因的失活、染色体移位、端粒酶的甲基化等共同作用的结果。基因水平检测可能提高患者肿瘤性质的诊断率。
中枢神经系统PNET的治疗以手术为基础,尽管切除程度和预后的关系还不十分肯定,但目前多数学者主张尽可能全切肿瘤,以及术后放疗、化疗、细胞免疫治疗的综合治疗模式。Majos等[10]在对中枢神经系统PNET治疗方案的前瞻性研究后认为,术后立即放疗再行化疗与接收放疗前化疗相比有较好的预后。陈忠平[11]建议治疗方案为,术后4~6周开始行全中枢神经系统放射治疗,分为全脑放疗及全脊髓放疗两部分进行,对发现椎管内PNET的患者应给予全脊柱加全脑方式的放射治疗以预防脑脊液转移。化疗方案很多,目前还没有确定标准的化疗方案,有报道[12]ICHT(长春新碱,顺铂,环磷酰胺,依托泊苷)方案效果较好。Fangusaro等[13]应用手术、化疗(ICHT方案)及自体造血干细胞移植的方法治疗43例儿童中枢神经系统PNET,5年无进展生存率及总体生存率可达到39%~49%。Citik等[14]在研究中发现,不论是初发还是复发PNET患者均可通过造血干细胞移植治疗获益。Sung等[15]报道,高剂量化疗能够延长高危患儿的生存期,在一定程度上,对复发的PNET也能起到延长生存期的作用;有ICHT、CAV(环磷酰胺+阿霉素+长春新碱)、CAVD(环磷酰胺+阿霉素+长春新碱+更生霉素)、大剂量顺铂、异环磷酰胺加二巯基乙醇硫酸钠法等。目前对其治疗方案有待进一步研究。本例患者行手术及放射治疗,未行化疗治疗,患者最终病情进展于术后9个月死亡。考虑若该患者术后配合化疗是否可能适量延长生存时间。
PNET的预后不佳,主要因为肿瘤易复发,是否有远处转移及年龄是影响预后的关键因素之一,是否手术切除或者全切及术后的辅助治疗也很重要。总之,中枢神经系统PNET是一种病程短、病死率高的疾病,生物学行为高度恶性,易复发、转移,预后极差。该肿瘤多见于30岁以下患者,生存率较低,基因突变及染色体移位是其可能的发病机制。其临床症状及影像学检查无特征性表现,病理学诊断仍是诊断的金标准。目前PNET的主要治疗方法主张以手术为主的综合治疗,术后辅以放疗及化疗等手段。因其发病年龄轻,病情进展快,预后极差,故需要加以重视,以防止误诊及延误治疗。
[1] Louis DN,Perry A,Reifenberger G,etal.The 2016 World Health Organization classification of tumors of the central nervous system:a summary[J].Acta Neuropathol,2016,131:803.
[2] 杨立,徐庆中.原始神经外胚层肿瘤[J].诊断病理学杂志,1997,4:226.
[3] Schmidt D,Herrmann C,Jürgens H,etal.Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma.A report from the Kiel Pediatric Tumor Registry[J].Cancer,1991,68:2251.
[4] Akihiro U,Hiroshi U,Shinsuke S,etal.A case of peripheral-type primitive neuroectodermal tumor arising in the dura mater at the frontal base[J].No To Shinkei,2004,56:237.
[5] Kim DG,Lee DY,Paek SH,etal.Supratentorial primitive neuroectodermal tumors in adults[J].J Neurooncol,2002,60:43.
[6] Pang C,Chang Q,Chung F,etal.Epigenetic inactivation of DLC-1 in supratentorial primitive neuroectodermal tumor[J].Hum Pathol,2005,36:36.
[7] Chang Q,Ng K.Different hypermethylation status of RASSF1A in medulloblastoma and supratentorial primitive neuroectodermal tumor[J].Zhonghua Bing Li Xue Za Zhi,2007,36:24.
[8] Antonelli M,Caltabiano R,Chiappetta CA,etal.Primary peripheral PNET/Ewing’s sarcoma arising in the meninges,confirmed by the presence of the rare translocation t(21;22) (q22;q12)[J].Neuropathology,2011,31:549.
[9] Minniti G,Tmish D,Ashley S,etal.Rjsk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma:update after an additional 10 years[J].J Clin Endocrinol Metab,2005,90:800.
[10] Carles M,Juli A,Carles A,etal.Adult primitive neuroectodermal tumor:proton MR spectroscopic findings with possible application for differential diagnosis[J].Radiology,2002,225:556.
[11] 陈忠平.神经系统肿瘤[M].北京:北京大学医学出版社,2009:477.
[12] Gutin PH.Cancer of the nervous system[J].Lippincott Williams & Wilkins,1998,42:305.
[13] Fangusaro J,Finlay J,Sposto R,etal.Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue(AuHCR)in young children with newly diagnosed supratentorial primitive neuro-ectodermal tumors(sPNETs):report of the Head Start.I and experience[J].Pediatr Blood Cancer,2008,50:312.
[14] Citik EC,Oguz A,Karadeniz C,etal.Primitive neuroectodermal tumor of the kidney in a child[J].Pediatr Hematol Oncol,2009,26:481.
[15] Sung KW,Yoo KH,Cho EJ,etal.High-dose chemotherapy and autologous stem cell rescue in children with newly diagnosed high-risk or relapsed medulloblastoma or supratentorial primitive neuroectodermal tumor[J].Pediatr Blood Cancer,2007,48:408.
(收稿2016-01-21 修回2016-03-26)
One case report of audlt supratentorial multiple primitive neuroectodermal tumor and literature review
HUChong-ling,LOUSi-long,MAODe-qiang,etal.
DepartmentofBrain,ChongqingCancerInstitute&Hospital&CancerCenter,Chongqing400030,China
Objective To explore the diagnosis and treatment of adult supratentorial primitive neuroectodermal tumor(PNET),to improve the early diagnostic rate,to improve the curative effect and prognosis.Methods Retrospective analysis of one audlt supratentorial multiple primitive neuroectodermal tumor who was in our hospital in March 2015,the patient imaging prompted intracranial multiple sites,and he did surgical operation treatment,most of the tumor resected,he accepted radiotheraphy according to postoperative pathology.And used “Pubmed”and“China net”to review relevant literature.Result Most of the tumor resected,the patient’s postoperative pathological prompted the central nervous system primitive neuroectodermal tumor,he accepted postoperative radiotherapy.By follow-up the condition of the patient worsed after postoperative four month and died after nine month.Retrieval results suggested:the reports of audlt supratentorial PNET from home and abroad,from 2005 to 2015,were 48 literature reports, including Pubmed searched 26 references,18 case reports of intracranial PNET,8 cases of retrospective analysis,“CNKI” search with 22 references,14 case reports of intracranial PNET,8 cases retrospective analysis,included 276 cases,however there had none case of the audlt supratentorial multiple PNET been reported.Conclusion The incidence rate of adult supratentorial PNET was low,high malignant degree,high rate of recurrence or metastasis,should be early diagnosis and timely surgical treatment,it maybe prolong patient life by treatment with radiation and chemotherapy.
primitive neuroectodermal tumor; supratentorial; audlt
10.3969/j.issn.1672-7770.2017.01.015
400030 重庆市肿瘤研究所/医院/ 癌症中心脑科
R739.43
D
1672-7770(2017)01-0055-04
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
国际放射医学核医学杂志(2021年10期)2021-02-28
浙江医学(2020年9期)2020-07-01
中国临床医学影像杂志(2019年4期)2019-06-18
浙江中西医结合杂志(2019年4期)2019-05-05
中国临床医学影像杂志(2019年1期)2019-04-25
浙江医学(2019年2期)2019-01-23
中国生殖健康(2019年9期)2019-01-07
畜牧兽医学报(2018年10期)2018-02-14
医学信息(2017年9期)2017-04-22
