
异羟肟酸对宫颈癌细胞生长抑制作用的研究
2016-12-29 19:14魏萍李苓张玮张小玲张婷婷盛修贵
特别健康·下半月 2016年12期
魏萍+李苓+张玮+张小玲+张婷婷+盛修贵
【中图分类号】R490.5 【文献标识码】A 【文章编号】2095-6851(2016)12-0-01
组蛋白去乙酰化酶抑制剂(HDI)是一类特异性抑制组蛋白去乙酰化酶、促进染色质解螺旋、增强其趋近性,从而调节基因转录的小分子化合物。体内、外研究证明组蛋白去乙酰化酶抑制剂可通过抑制肿瘤细胞增殖、诱导凋亡、抑制肿瘤血管生成及转移、降低肿瘤侵袭力等机制发挥抗肿瘤作用,与放射联合还有放射增敏作用[1]。
近来研究表明, 异羟肟酸有组蛋白去乙酰化酶(histone deacetylase , HDAC)抑制剂活性, 可抑制多种肿瘤细胞生长。异羟肟酸被广泛称为低毒性HDAC抑制剂。Tambunan US[2]等研究提出了利用苯三唑对异羟肟酸修改,获得一个更好的抑制剂。本研究观察异羟肟酸对宫颈癌Hela 和Siha 细胞株生长和细胞周期的影响及p53mRNA 和蛋白表达改变。
1.材料与方法
1.1 材料
异羟肟酸购自Sigma 公司,用PBS 稀释成500mmol/L, 过滤分装,贮藏于- 20℃。P53抗体购自Santa Cruz 公司,为鼠抗人单抗。RPMI-1640 (Gibco 公司)。TRIZOL 试剂购自Invitrogen 公司。新生牛血清(杭州四季青)。FACSort 流式细胞仪为美国Becton Dickson 公司产品。四甲基偶氮唑蓝(MTT)、二甲基亚砜(DMSO)(Sigma);AnnexinV-FITC 凋亡和细胞周期分析试剂盒为Becton Dickinson 公司产品。
1.2 实验方法
1.2.1 细胞和细胞培养
Hela 和Siha 细胞株由本科室保存。细胞在5% CO2、37℃条件下,用含10% 新生牛血清、青霉素(100U/ml)、链霉素(100U/ml)的RPMI-1640 培养液培养传代。
1.2.2 MTT 实验检测生长抑制率
取对数生长期宫颈癌细胞,按细胞浓度为5×104/ 孔接种于96 孔板。37℃、5%CO2 饱和湿度培养24h 后, 分别加入不同浓度SAHA(0, 1.2, 2.4, 5.0 mM) 的培养基,每孔200μl,对照组不做处理。分别于加药后0、1、2、3和4天,每孔加入MTT 溶液(5mg/ml)20μl, 37℃避光培养4h。彻底吸去MTT 溶液,加入二甲基亚砜150μl/孔, 震荡10min, 酶标仪490nm激发波长检测吸光度。计算细胞的生长抑制率。细胞生长抑制率(%)=(1- 实验组吸光度值/对照组吸光度值)×100%。每次实验设3 复孔, 实验重复3 次。
1.2.3 流式细胞仪(FCM)检测细胞凋亡和细胞周期
Hela 细胞和Siha 细胞分别用含(0, 1.2, 2.4, 5.0 mM) SAHA的培养基培养48h 。胰酶消化成单细胞悬液, 1 000r/min 离心5min收集细胞, 加0.01mol/L PBS 洗涤离心2 次, 用4℃75% 乙醇固定过夜。1 000r/ min 离心5min , 去掉乙醇固定液, PBS 洗涤2 次。加入PI 染液1ml(50μg/mlPI、20μg/ml RNase、0.1% Triton-100), 4℃孵育30min , FCM检测凋亡细胞率。
1.2.4 RT- PCR 检测p53表达
分别收集用含(0, 1.2, 2.4, 5.0 m M) SAHA处理48h的细胞。用TRIZOL 提取细胞总mRNA, 参照说明书进行。提取的mRNA 用紫外分光光度计检测RNA的纯度和浓度(A260 /A280>1.8)。取2μg mR-NA 为模板, 应用通用引物逆转录合成cDNA, 以cDNA 为模板进行PCR 扩增。P53 引物: 正义链5′-cctcttcggcccagtggac-3 ′, 反义链5′-ccgttttcgaccctgagag-3 ′,退火温度60℃, 共28 个循环, 扩增产物369bp ;β-actin 引物: 正义链5′-ggcatcgtgatggactccg-3 ′,反义链5′-gctggaaggtggacagcga-3 ′, 退火温度62℃, 共28个循环, 扩增产物594bp 。扩增条件为:94 ℃预变性3 min; 94 ℃ 40 s, 56 ℃ 40 s, 72 ℃ 1min, 30 个循环; 72 ℃延伸7 min。取PCR 产物5 μl在1.5%琼脂糖凝胶上电泳( 100 V, 30 min) 。凝胶成像系统成像, 经薄层扫描求得每个待测产物与β-actin 产物光密度之比, 进行半定量分析。 PCR 产物在1.5% 的琼脂糖凝胶上电泳, 电泳结果用英国UVP 公司GDS8000 型全自动图像分析系统处理。
1.6 Western blot 分析p53蛋白表达水平 分别收集0, 1.2, 2.4, 5.0 m M SAHA 处理72 h 的细胞, 提取蛋白质, 采用Bradford 法定量蛋白质,取50μg蛋白进行SDS- PAGE 凝胶电泳。然后电转膜到硝酸纤维素膜上。室温下摇动封闭4h。TBS-T洗涤3 次, 加一抗(p53按1 ∶100 稀释)在4 ℃下过夜。室温下洗膜后加入辣根过氧化物酶偶联兔抗小鼠IgG 抗体, 洗去未结合的二抗, 滴加化学发光剂ECL 进行发光显影。TBS- T 洗涤3 次,再用二抗(1 ∶5 000 稀释的辣根过氧化物酶标记的羊抗鼠IgG)振荡孵育2h 。采用化学发光法( ECL)显影。
1.3 统计学处理
计量资料用均数±标准差(±s) 表示,用SPSS11.5 软件进行统计,数据比较采用t 检验,单变量多组资料之间比较采用单因素方差分析, 以P<0.05 为差异具有统计学意义。
2.结果
2.1 异羟肟酸抑制细胞生长
MTT 检测结果显示, 异羟肟酸显著抑制Hela 和Siha 细胞生长,呈时间和浓度依赖性( 见图1A、1B )。不同浓度( 0~5 mmol /L)SAHA处理Hela 和Siha 细胞可抑制细胞生长, 差异有统计学意义( F=4.87, P<0.01);SAHA处理不同时间( 0 h~96 h) 可抑制细胞生长, 差异有统计学意义( F=3.78, P<0.01)。经直线回归方程计算,异羟肟酸作用48、72、96 h 时IC50 值分别为14.08、6.78、4.25 mmol /L。
2.2 异羟肟酸对细胞周期的抑制
异羟肟酸可显著影响宫颈癌Hela 和Siha 细胞的周期分布,使Hela 和Siha 细胞G0/G1 期比例显著增加(P =0.001)( 见表1 ) 。异羟肟酸降低Hela 细胞S 期( P=0.052 ) 和G2/M 期( P =0.059 ) 比例, 但差异无显著性。异羟肟酸降低Siha 细胞S 期( P =0.004 ) 和G2/M 期( P=0.007 ) 比例, 差异有显著性。
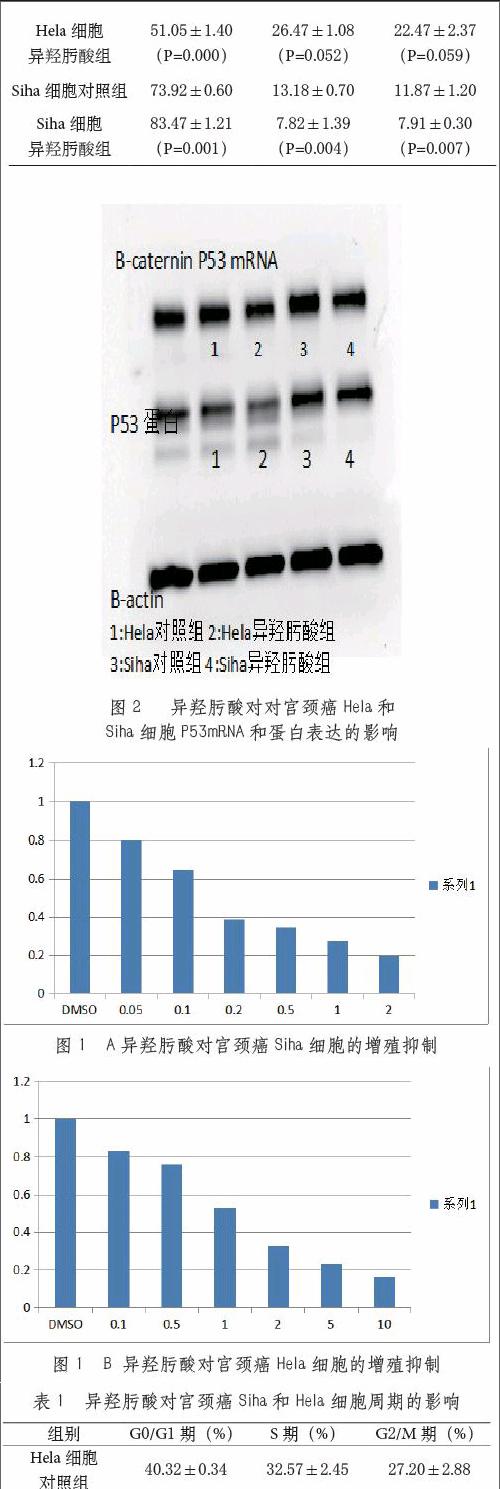
2.3 异羟肟酸上调p53表达
RT- PCR 检测显示异羟肟酸上调Hela 和Siha细胞的p53mRNA 表达 。Westernblot 检测提示异羟肟酸促进Hela 和Siha 细胞的p53蛋白表达( 见图2) 。
3.讨论
现有大量研究表明,癌症的发生与表观遗传学有着密切的关系,主要包括组蛋白乙酰化、磷酸化、泛素化及DNA甲基化等,其在基因转录调节中起着重要的作用。Zhang Y[3]等在2004年发现组蛋白脱乙酰酶抑制剂能增强人鳞癌细胞放疗的敏感性。为了进行基因表达,细胞必须在组蛋白周围调控DNA的螺旋和非螺旋,而这一过程需在组蛋白乙酰化酶的协助下方能完成,故HDAC抑制剂可引起组蛋白超乙酰化,从而影响细胞的基因表达。HDACs 不仅仅能够催化组蛋白的去乙酰化,而且能够催化其他一些重要蛋白( 如Hsp90、Tubulin 等) 和转录因子( p53、STAT1 等) 的乙酰化. HDAC 在基因转录中起重要作用[4];其可与某些转录因子( 如E2F、Stat3、p53、NF-_B、TFIIE 及Rb 蛋白) 发生相互作用,从而调节基因转录[5]。
研究表明p53蛋白羧基端乙酰化能抑制乙酰化赖氨酸与泛素酶的结合并由此引起的降解[6], 而这种单泛素化降解能促进p53蛋白的核转运。因此p53羧基端乙酰化可稳定p53在核内停滞并影响其在线粒体中的功能表达。同时,在正常情况下,乙酰化p53在细胞中的比例很低。反映出在体内具有强去乙酰化作用[7-9]。早期研究显示在多种肿瘤细胞中,p53羧基端乙酰化状态能调节转录凋亡前效应[10-15]。
Xing J[16]等研究不同浓度SAHA 联合顺铂(DDP)治疗人类宫颈癌SiHa细胞,抑制和细胞凋亡率和细胞周期。SiHa细胞处理后20%的IC50SAHA24小时,经历了各种剂量的辐射处理。S发现AHA在化疗时通过上调p21和Bax促进SiHa信使rna和蛋白质的细胞凋亡,导致细胞周期阻滞于G0 / G1期。低剂量的SAHA在放疗时通过Bax上调和Ku70下调促进SiHa细胞凋亡和抑制细胞修复。
本研究证实异羟肟酸抑制人宫颈癌Hela 和Siha细胞生长,可呈药物依赖性地抑制细胞增殖、诱导细胞凋亡。阻滞于G0-G1期的细胞明显增多, P53水平显著上升。异羟肟酸使Hela 和Siha细胞G0/G1 期细胞比例显著增加, 推测异羟肟酸主要通过影响细胞周期, 使细胞阻滞于G0/G1 期, 从而抑制宫颈癌细胞生长。我们观察到, 异羟肟酸对Hela 细胞的生长抑制强于对Siha 细胞的生长抑制。这可能因为Hela 细胞G0/G1 期比例明显低于Siha细胞, Hela 细胞中有更多细胞处于S 期和G2/M 期, 推测异羟肟酸可能对增殖旺盛细胞的生长抑制作用更强。在宫颈癌细胞株中, HDAC抑制剂可经P53 途径诱导肿瘤细胞凋亡[17]。HDAC抑制剂可增加P53的转录,上调P53基因的表达,阻止P53蛋白被E6降解[18]。本实验中观察到异羟肟酸上调p53表达, 引起G0/G1 期阻滞, 可见细胞凋亡。
新型HDAC 抑制剂在小剂量、低氧浓度情况下可诱导肿瘤细胞分化、选择性凋亡。HDAC 抑制剂作为一种新型的靶向抗癌药,正成为药物研究的新热点,临床应用前景广泛。这类抑制剂可以靶向性地使肿瘤细胞出现生长停滞、分化、凋亡,对正常细胞无毒性,而且其抗肿瘤谱广,实用性较强,临床潜力巨大。
参考文献
[1]Ropero S, Esteller M. The role of histone deacetylases(HDACs) in human cancer[J]. Mol Oncol, 2007,1(1):19-25.
[2]Tambunan US1, Bramantya N, Parikesit AA et,al..In silico modification of suberoylanilide hydroxamic acid (SAHA) as potential inhibitor for class II histone deacetylase (HDAC)[J]. BMC Bioinformatics 2011.
[3]Zhang Y,Jung M,Dritschilo A,et al.Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors[J].Radiat Res.2004 Jun;161(6):667-74.
[4]Lin HY, Chen CS, Lin SP, et al. Tar-geting histone deacetylasein cancer therapy[J]. Med Res Rev, 2006, 26(4): 397-413.
[5] ZhangY,JungM,DritschiloA, et al. Enhancementof radi2ation sensitivityof human squamous carcinoma cells byhistone deacetylase inhibitors[J]. Radiat Res, 2004, 161:6672674
[6] Grossman SR, Deato ME, Brignone C,et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300[J]. Science, 2003,300(5617): 342-344.
[7] Luo J, Li M,Tang Y, et al. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo[J]. Proc Natl Acad Sci U S A, 2004,101(8): 2259-2264.
[8] Li M, Luo J, Brooks CL, et al. Acetylation of p53 inhibits its ubiquitination by Mdm2[J]. J Biol Chem, 2002, 277(52): 50607-50611.
[9]Zhao Y, Lu S,Wu L, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1)[J]. Mol Cell Biol, 2006, 26(7): 2782-2790.
[10] Zhang S, Cao HJ, Davis FB, et al. Oestrogen inhibits resveratrol-induced post-translational modification of p53 and apoptosis in breast cancer cells[J]. Br J Cancer, 2004, 91(1): 178-185.
[11]Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress[J]. Cell, 2001, 107(2):137-148.
[12] Fu M, Wang C, Zang X, et al. Acetylation of nuclear receptors in cellular growth and apoptosis[J]. Biochem Pharmacol, 2004.68(6): 1199-1208.
[13] Vaghefi H, Neet KE. Deacetylation of p53 after nerve growth factor treatment in PC12 cells as a post-translational modification mechanism of neurotrophin-induced tumor suppressor activation[J]. Oncogene,2004,23(49): 8078-8087.
[14] Imanishi R, Ohtsuru A, Iwamstsu M, et al. A histone deacetylase inhibitor enhances killing of undifferentiated thyroid carcinoma cells by p53 gene therapy[J]. J Clin Endocrinol Metab, 2002, 87(10): 4821-4824.
[15] Luo J, Su F, Chen D, et al. Deacetylation of p53 modulates its effect on cell growth and apoptosis[J]. Nature, 2000,408(6810): 377-381.
[16]Xing J, Wang H, Xu S, et al.Sensitization of suberoylanilide hydroxamic acid (SAHA) on chemoradiation for human cervical cancer cells and its mechanism[J].Eur J Gynaecol Oncol[ J ]. 2015,36(2):117-22.
[17] Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer[J]. Mol Oncol, 2007, 1(1): 19-25.
[18] Cruz-Hernandez E, et al. The effects of DNA methylation and histone deacetylase inhibitors on human papillomavirus early gene expression in cervical cancer, an in vitro and clinical study[J]. Virol J. 2007,16(2):216-22..
猜你喜欢
中国医药科学(2022年9期)2022-07-13
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
中国药学药品知识仓库(2022年7期)2022-05-10
医学概论(2022年4期)2022-04-24
中国药房(2022年7期)2022-04-14
三农资讯半月报(2021年1期)2021-01-27
保健与生活(2019年3期)2019-08-01
中学生物学(2016年10期)2016-11-19
现代养生·下半月(2015年8期)2015-11-16
