
三种气体信号分子在动脉粥样硬化发生发展中的相互作用
2016-12-24 17:02,*,
中南医学科学杂志 2016年4期
,*,
(1.南华大学附属南华医院,湖南 衡阳 421001;2.南华大学心血管病研究所,动脉硬化学湖南省重点实验室)
·文献综述·
三种气体信号分子在动脉粥样硬化发生发展中的相互作用
肖文超1,马小峰1*,姜志胜2*
(1.南华大学附属南华医院,湖南 衡阳 421001;2.南华大学心血管病研究所,动脉硬化学湖南省重点实验室)
动脉粥样硬化是冠心病、脑梗死、外周血管病等众多心脑血管疾病的主要病理基础。目前发现的三种气体信号分子一氧化氮、一氧化碳与硫化氢在动脉粥样硬化中有调节血管张力,抗血小板在内皮细胞中的粘附,抑制血管平滑肌细胞的增殖和迁移等作用。在动脉粥样硬化形成的过程中,它们两两之间起协同或代偿作用,抑制动脉粥样硬化的发生发展。
一氧化氮; 一氧化碳; 硫化氢; 动脉粥样硬化
动脉粥样硬化(atherosclerosis,As)是一种主要累及主动脉及中动脉的病理过程,包括内皮细胞功能障碍,血管平滑肌细胞的增殖和迁移,炎症细胞募集,脂质积聚和血栓形成等[1],是多种心脑血管疾病共同的病理基础。上个世纪八十年代,有人发现气体信号分子一氧化氮(nitricoxide,NO)具有舒张血管、抑制平滑肌细胞增殖、抗血栓、等生物学效应,在抗动脉粥样硬化形成过程中发挥了很重要的作用。随后,又相继发现了一氧化碳(carbon monoxide,CO)和硫化氢(hydrogensulfide,H2S)这两种新的气体信号分子,并发现他们能内源性产生具有广泛的生物学效应,在动脉粥样硬化的发生发展中也扮演重要角色。
近年来,越来越多的研究关注于三种气体信号分子NO、CO和H2S与动脉粥样硬化之间的关系。尤其是他们两两之间的“cross talk”(见图1)——气体信号分子间的相互影响和相互调节作用,这对于进一步阐明气体信号分子在动脉粥样硬化中的作用机制有重要意义。
1 三种气体信号分子的生成与功能
1.1 NO NO是体内最早发现的气体信号分子,由NO合成酶(Nitric oxide synthase,NOS)蛋白家族催化L-精氨酸(L-Arginine)产生。NOS有三种同工酶,其中内皮型NOS(endothelial NOS,eNOS)与神经元型NOS(neuronal NOS,nNOS)为结构型NOS(cNOS),第三种为诱导型NOS(inducible NOS,iNOS)。nNOS主要表达于神经系统,eNOS主要表达于血管内皮细胞、血小板,而iNOS主要表达于血管平滑肌细胞、巨噬细胞以及中性粒细胞等。
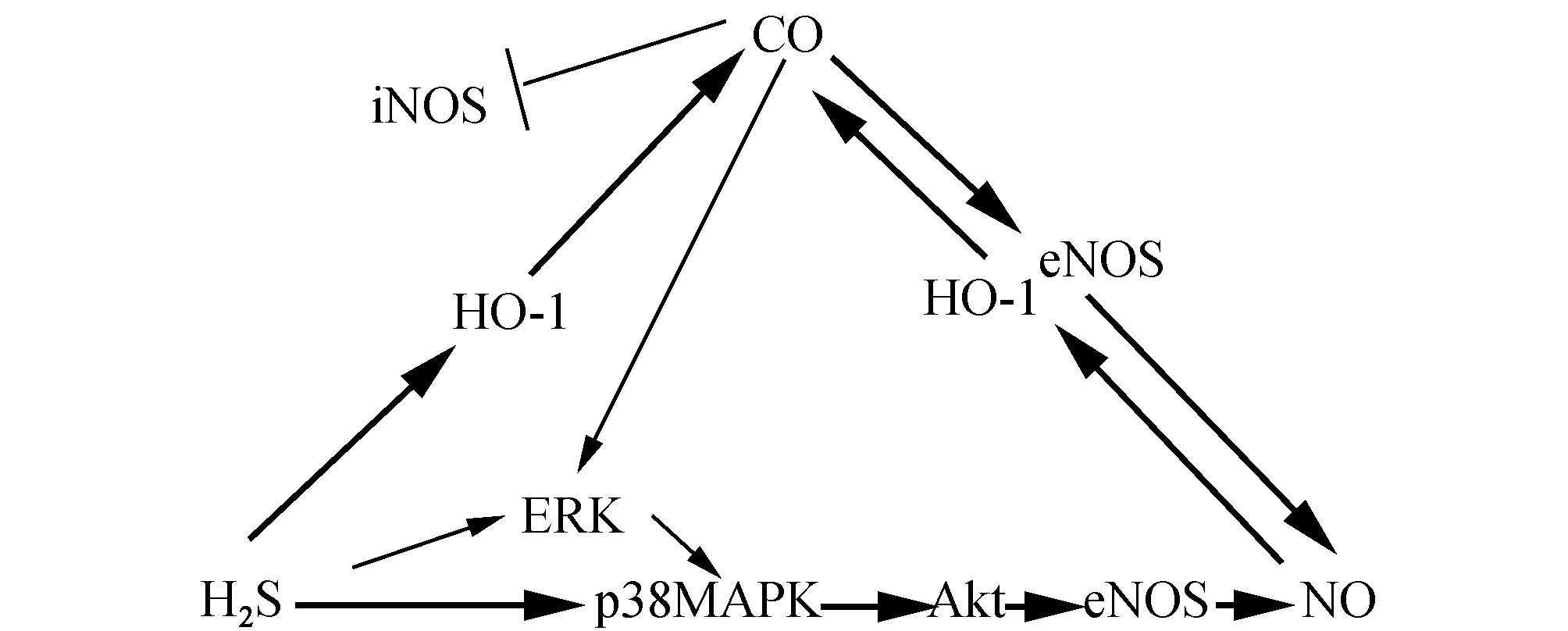
图1 三种气体信号分子间的“cross talk”
在动物实验中,iNOS[2]表现出促动脉粥样硬化作用,而nNOS与eNOS[3]表现出抗动脉粥样硬化作用。NO发挥抗动脉粥样硬化作用的途径包括:调节血管张力,抗氧化应激,抑制粘附分子在内皮细胞中粘附和聚集,抗血栓形成,抑制平滑肌细胞的迁移和增殖等(见表1)。
1.2 CO 体内绝大部分CO的主要来源是血红素,并受血红素加氧酶(hemeoxygenase,HO)调控。体内被确认的HO同工酶有三种,氧化应激诱导的HO-1和结构型 HO-2,HO-3。在动脉粥样硬化病变过程中,HO-1被诱导激活,产生大量CO。CO可通过变构和激活促环鸟苷酸(cGMP) 的生成,舒张血管平滑肌细胞,参与血压调控[4],从而在心血管系统和抗动脉粥样硬化过程中起积极作用。CO与NO的作用类似,可作为内皮依赖型血管舒张因子,CO不仅能调控血管舒缩功能,并且能抑制单核细胞的聚集和抗血小板粘附,抗脂质沉积,抑制血管平滑肌细胞的增殖和迁移(见表1)。
表1三种气体信号分子的生成及其在抗As中的作用
在TNF-α诱导的血管内皮细胞(vascular endothelial cells,VECs)中,CO供体CORM-A1能抑制Nox4介导的活性氧(Reactive oxygen species,ROS)生成,通过Akt与MAPKs 信号通路减轻内皮细胞的损伤,提高细胞存活率[5]。相反,用HO抑制剂预处理血管平滑肌细胞后,ROS生成增多[6]。CORM-2可以降低血清C反应蛋白和氧化低密度脂蛋白,并且抑制脂质在主动脉中的沉积[7]。 CORM-2协同羟基酪醇(HT) 时几乎能完全抑制TNFα介导的IκBα和NFκBp65磷酸化[8]。
1.3 H2S H2S以前被认为是一种有毒的环境污染物,近些年的研究表明,它是一种新型的气体信号分子,在人体内拥有多种生物学效应。内源性H2S在哺乳动物细胞中通过不同的方式产生。其合成大部分通过酶催化,包括使用L-半胱氨酸为底物的胱硫醚γ裂解酶(cystathionine-γ-lyase,CSE)及胱硫醚β合酶(cystathionine-β-synthase,CBS)或使用3-巯基丙酮酸作为底物的3-巯基丙酮酸硫转移酶(3-mercaptopyruvate sulfurtransferase,3-MST)催化生成。 CBS和3-MST主要表达于脑部,而CSE大量表达于组织和细胞中,包括肝、肾、心脏、血管平滑肌细胞和内皮细胞。H2S的抗动脉粥样硬化作用主要表现在促血管平滑肌细胞增殖、抑制其凋亡,抑制ROS的产生、黏附分子的聚集和泡沫细胞的形成等[9](见表1)。
补充外源性H2S(NaHS或Na2S),H2S生成底物半胱氨酸或过表达CSE提高内源性H2S的生成都能降低氧化低密度脂蛋白(ox-LDL)预处理的巨噬细胞中肿瘤坏死因子α (TNF-α)的产生,同时减少细胞黏附分子-1 (ICAM-1)的生成,减弱巨噬细胞对血管内皮细胞的黏附作用[10],明显抑制COX2,iNOS的生成。有研究表明,21三体综合征患者的动脉粥样硬化病理进程显著低于正常人,而在他们体内,H2S水平显著增高。
2 三种气体信号分子在动脉粥样硬化中的“cross talk”
2.1 CO和H2S的“cross talk” 生理情况下,HO-1/CO通路与CSE/H2S通路之间存在相互竞争性抑制关系。在主动脉平滑肌细胞(arterial smooth muscle cell,ASMC)中,使用H2S抑制剂PPG能提高CO水平。另一方面,内源性CO抑制剂ZnPP能显著提升CSE的表达和H2S水平。Yamamoto T等[11]的研究表明,CO可能是H2S生成过程中CBS的抑制剂。接着,Takayuki Morikawa等[12]又证明了HO-2/CO在脑血管中抑制CBS/H2S通路以及H2S的生成,在血管内皮细胞中,CBS生成的H2S也能被HO-2生成的CO所抑制。
而在病理情况下,这两种气体信号分子却表现出完全不一样的特性。在内毒素休克导致的急性肺损伤(acute lung injury,ALI)中,提高H2S水平导致HO-1/CO信号通路的活性的提高。外源性H2S能减缓缺氧性肺动脉高压的病理进程,降低肺动脉压,同时上调HO-1、HO-1 mRNA 的表达与CO的生成,而内源性H2S抑制剂炔丙基甘氨酸(PPG)加重了HPH的病理过程[13]。在LPS诱导的RAW264.7 巨噬细胞中,H2S通过上调HO-1/CO通路,抑制iNOS的生成,减轻了iNOS对内皮细胞的损害作用,并抑制炎性反应[14]。当使用HO抑制剂下调CO后,H2S对iNOS的抑制作用减弱。其作用机制是使NF-κB失活[15]。在动脉粥样硬化进程中,CO 与H2S 在抗损伤时主要起协同作用,而H2S也许是抗损伤的内源性补偿。CO与H2S相互之间具体调控的机制仍不清楚,还需要进一步的探索研究。
2.2 NO和H2S的“cross talk” 生理浓度的H2S能刺激生成NO,Predmore等[16]的研究证明,使用低浓度Na2S (150 μmol/L)处理牛主动脉内皮细胞能刺激NO的生成。其可能的机制是H2S激活了eNOS的上游通路。在内皮细胞中,H2S通过一系列磷酸化作用激活eNOS。首先,H2S磷酸化激活p38 MAPK,随即磷酸化激活下游Akt/eNOS信号通路,从而增加NO的生成。抑制p38 MAPK后,H2S诱导的Akt磷酸化即失效,也无法增加NO。同时,H2S也能时间依赖性磷酸化上调ERK,它能通过p38 MAPK的激活增加NO的产生,但使用ERK抑制剂时并不会影响H2S诱导的NO的形成[17]。一种新型的特异性荧光分子探针FA-OMe同时证实H2S可以通过磷酸化丝氨酸1177位点激活AMPK/Akt信号通路从而提高NO产量,同时通过提高蛋白质巯基亚硝基化以提高NO的生物利用率[18]。有趣的是,高浓度H2S反而表现出完全相反的特性。使用300~3 000 μmol/L的NaHS处理小鼠及大鼠主动脉,能明显抑制eNOS的生成,从而抑制NO的形成[19]。
在血管内皮细胞的激活、增殖、迁移及新的血管和血管网的形成过程中,H2S与NO两者相互影响又相互独立。他们能独立或协同促进血管新生,刺激内皮细胞的增殖或迁移。而H2S的促血管生成作用不能被NO抑制剂完全抑制。在促血管生成过程中,NO可能为关键因素。在eNOS-/-敲除小鼠中,H2S治疗能再通75% 的缺血后肢,并且在使用NOS抑制剂L-NAME后仍能发挥作用,却在使用NO清除剂后失效[20]。H2S促进血管重建是通过增加HIF-1α 与VEGF的表达和活性,同时促进 NOS 的表达和提高NO的生物利用率[20]。同样的,NO诱导的血管再生同样需要H2S 的产生。使用shRNA完全沉默CSE以后,血管生成被完全抑制,加入NO供体也无法解除这种抑制。H2S促NO生成的机制是通过Ser493位点磷酸化激活Akt通路,最终激活eNOS。此外,H2S 呈浓度依赖性阻止cGMP 的失活从而提升cGMP 水平,激活下游PKG通路。CSE/H2S在氧化应激中的保护作用依赖于eNOS/NO[21]。H2S预处理并不能使eNOS磷酸化失活突变小鼠(S1179A)免于细胞损伤和氧化损伤[21],其潜在的治疗功能在于通过多位点磷酸化eNOS/NO通路以提高NO的产量和生物利用度,以及提高cGMP水平,在抗动脉粥样硬化斑块形成时协同NO作用[21]。
H2S对血管的舒张作用同样需要eNOS/NO参与[22]。L-NAME诱导的高血压大鼠中,外源性H2S可通过KATP通道上调NO的表达和NO的生物利用率,改善eNOS功能失调[22]。用NaHS 30 μmol/L处理主动脉环15 min后,NO对血管舒张的调节作用更强。CSE的增加亦能逆转NO抑制导致的高血压[23]。另有研究表明,野生型小鼠胸主动脉比eNOS-/-敲除小鼠对NaHS的反应更敏感。在肥胖小鼠中,H2S介导瘦素诱血管舒张,能代偿肥胖小鼠的NO缺陷[24]。H2S同样通过NO的产生,能减轻在振荡剪切应力下导致的单核细胞对内皮细胞的黏附作用,阻止ICAM-1 的释放[25],它通过对Akt的磷酸化作用提高eNOS水平[25]。
事实上,探索H2S与NO两者相互之间的影响时,这两种气体信号分子药物供体的特异性可能对实验过程及结果有影响。但总的来说,在动脉粥样硬化的病理进程中,两者相互代偿并起协同作用。当一方受到损害时,提高另一方的含量能减少受损,起到抗动脉粥样硬化作用。并且,在很多情况下,H2S发挥其抗动脉粥样硬化作用是NO依赖的。
2.3 CO和NO的“cross talk” 生理情况下,对血管内皮细胞,NO和CO具有类似的生理功能,都是通过cGMP起作用。但由于NO对cGMP亲和力更强,是CO的50倍,所以NO发挥了更重要的生理作用。它们也通过Ca2+通道参与血管舒张[26]。在动脉粥样硬化血管组织中,cNOS的活性和NO水平显著降低,而HO-1的表达和活性以及CO的产量上升[27],这说明 HO-1/CO 信号通路起到代偿或与cNOS/NO相同的作用。
NO能被CO诱导,CO抑制动脉粥样硬化可通过iNOS的抑制同时促进eNOS的生成[28]。在血管内皮细胞中,CORM-2联合羟基酪醇显著提升eNOS的活性,并增加了NO的产量,这一途径是通过增加eNOS的磷酸化水平、激活ERK来实现[8]。HO-1/CO可抑制iNOS的生成,可能的原因为:①iNOS为HO-1的底物,HO-1加速其分解;②由于CO与iNOS结合,使iNOS失活;③HO-1加速血红素的分解,减少iNOS的产生;④通过失活NF-κB来实现[15],并通过这一途径抑制巨噬细胞炎性反应[29]。在兔主动脉斑块中发现,使用了HO-1抑制剂锌原卟啉IX (Znpp-IX)后,iNOS水平显著上升,同时斑块面积明显大于对照组。这说明HO-1/CO能抑制iNOS的活性从而减轻动脉粥样硬化损伤[27]。在血管内皮细胞和血管平滑肌细胞中,提高NO能诱导HO-1的表达,提高作为NO非依赖性的血管舒张因子CO的水平。
然而,单独使用CO供体会减弱NO供体的血管舒张作用,它意味着CORM-2清除了生成的NO[30]。这种矛盾的实验结果可能跟实验时使用的供体和用量有关。
3 结 语
随着对三个气体信号分子NO、CO、H2S研究的深入,它们在动脉粥样硬化发生发展过程中两两或三者间的相互影响、相互调节引起了越来越多的关注。不同供体、不同用量和不同状态下,气体信号分子在动脉粥样硬化中的“cross talk”甚至呈现出完全相反的效应。弄清楚气体信号分子之间的关系,气体量效在动脉粥样硬化中的影响和作用,有助于抗动脉粥样硬化药物的开发和配合使用,对动脉粥样硬化的临床研究有重要的指导意义。
[1] van Lammeren GW,den Ruijter HM,Vrijenhoek JE,et al.Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery[J].Circulation,2014,129(22):2269-2276.
[2] Kuhlencordt PJ,Chen J,Han F,et al.Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice[J].Circulation,2001,103(25):3099-3104.
[3] Ponnuswamy P,Schrottle A,Ostermeier E,et al.eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice[J].PLoS One,2012,7(1):e30193.
[4] Ndisang JF,Tabien HE,Wang R.Carbon monoxide and hypertension[J].J Hypertens,2004,22(6):1057-1074.
[5] Basuroy S,Tcheranova D,Bhattacharya S,et al.Nox4 NADPH oxidase-derived reactive oxygen species,via endogenous carbon monoxide,promote survival of brain endothelial cells during TNF-alpha-induced apoptosis[J].Am J Physiol Cell Physiol,2011,300(2):C256-C265.
[6] Moraes JA,Barcellos-de-Souza P,Rodrigues G,et al.Heme modulates smooth muscle cell proliferation and migration via NADPH oxidase:a counter-regulatory role for heme oxygenase system[J].Atherosclerosis,2012,224(2):394-400.
[7] Hou M,Wang P,Zhao L,et al.Effect of CORM-2 on atherosclerosis in experimental periodontitis of rats[J].Shanghai Kou Qiang Yi Xue,2014,23(5):531-538.
[8] Zrelli H,Wu C W,Zghonda N,et al.Combined treatment of hydroxytyrosol with carbon monoxide-releasing molecule-2 prevents TNF alpha-induced vascular endothelial cell dysfunction through NO production with subsequent NFkappaB inactivation[J].Biomed Res Int,2013,2013:912431.
[9] Mani S,Untereiner A,Wu L,et al.Hydrogen sulfide and the pathogenesis of atherosclerosis[J].Antioxid Redox Signal,2014,20(5):805-817.
[10] Wang XH,Wang F,You SJ,et al.Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage[J].Cell Signal,2013,25(11):2255-2262.
[11] Yamamoto T,Takano N,Ishiwata K,et al.Carbon monoxide stimulates global protein methylation via its inhibitory action on cystathionine beta-synthase[J].J Clin Biochem Nutr,2011,48(1):96-100.
[12] Morikawa T,Kajimura M,Nakamura T,et al.Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway[J].Proc Natl Acad Sci U S A,2012,109(4):1293-1298.
[13] Li XH,Du JB,Ding YG,et al.Impact of hydrogen sulfide donor on experimental pulmonary hypertension induced by high pulmonary flow and endogenous carbon monoxide/heme oxygenase pathway[J].Beijing Da Xue Xue Bao,2006,38(2):135-139.
[14] Zhang S,Wu T,Xia T,et al.Regulation of the Heme Oxygenase-1/carbon monoxide system by hydrogen sulfide in murine coxsackievirus B3-induced myocarditis[J].Cell Mol Biol (Noisy-le-grand),2015,61(2):69-73.
[15] Oh GS,Pae HO,Lee BS,et al.Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide[J].Free Radic Biol Med,2006,41(1):106-119.
[16] Predmore BL,Julian D,Cardounel AJ.Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism[J].Front Physiol,2011,2:104.
[17] Altaany Z,Yang G,Wang R.Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells[J].J Cell Mol Med,2013,17(7):879-888.
[18] Chen PH,Fu YS,Wang YM,et al.Hydrogen sulfide increases nitric oxide production and subsequent S-nitrosylation in endothelial cells[J].Scientific World J,2014,2014:480387.
[19] Kubo S,Doe I,Kurokawa Y,et al.Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide:contribution to dual modulation of vascular tension[J].Toxicology,2007,232(1-2):138-146.
[20] Bir SC,Kolluru GK,McCarthy P,et al.Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis[J].J Am Heart Assoc,2012,1(5):e4093.
[21] King AL,Polhemus DJ,Bhushan S,et al.Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent[J].Proc Natl Acad Sci U S A,2014,111(8):3182-3187.
[22] Ji W,Liu S,Dai J,et al.Hydrogen sulfide defends against the cardiovascular risk of Nw-nitro-L-argininemethyl ester-induced hypertension in rats via the nitric oxide/endothelial nitric oxide synthase pathway[J].Chin Med J (Engl),2014,127(21):3751-3757.
[23] Oosterhuis N R,Frenay A R,Wesseling S,et al.DL-propargylglycine reduces blood pressure and renal injury but increases kidney weight in angiotensin-II infused rats[J].Nitric Oxide,2015,49:56-66.
[24] Jamroz-Wisniewska A,Gertler A,Solomon G,et al.Leptin-induced endothelium-dependent vasorelaxation of peripheral arteries in lean and obese rats:role of nitric oxide and hydrogen sulfide[J].PLoS One,2014,9(1):e86744.
[25] Go YM,Lee HR,Park H.H(2)S inhibits oscillatory shear stress-induced monocyte binding to endothelial cells via nitric oxide production[J].Mol Cells,2012,34(5):449-455.
[26] Huo L,Zhang J,Qu Z,et al.Vasorelaxant effects of Shunaoxin pill are mediated by NO/cGMP pathway,HO/CO pathway and calcium channel blockade in isolated rat thoracic aorta[J].J Ethnopharmacol,2015,173:352-360.
[27] Liu DN,Fang Y,Wu LR,et al.Effect of the haeme oxygenase-1/endogenous carbon monoxide system on atherosclerotic plaque formation in rabbits[J].Cardiovasc J Afr,2010,21(5):257-262.
[28] Wen Z,Liu Y,Li F,et al.Low dose of carbon monoxide intraperitoneal injection provides potent protection against GalN/LPS-induced acute liver injury in mice[J].J Appl Toxicol,2013,33(12):1424-1432.
[29] Liu XH,Wang XL,Xin H,et al.Induction of Heme Oxygenase-1 by Sodium 9-Hydroxyltanshinone IIA Sulfonate Derivative Contributes to Inhibit LPS-Mediated Inflammatory Response in Macrophages[J].Cell Physiol Biochem,2015,36(4):1316-1330.
[30] Marazioti A,Bucci M,Coletta C,et al.Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules[J].Arterioscler Thromb Vasc Biol,2011,31(11):2570-2576.
10.15972/j.cnki.43-1509/r.2016.04.028
2016-03-04;
2016-06-24
国家自然科学基金资助(30971169).
*通讯作者,E-mail:1507251097@qq.com;578151825@qq.com.
R541.4
A
蒋湘莲)
猜你喜欢
今日农业(2022年14期)2022-09-15
世界科学技术-中医药现代化(2022年3期)2022-08-22
波谱学杂志(2022年1期)2022-03-15
现代临床医学(2021年5期)2021-11-02
中国眼镜科技杂志(2019年9期)2019-11-11
模具制造(2019年7期)2019-09-25
天津医科大学学报(2019年6期)2019-08-13
天津医科大学学报(2019年6期)2019-08-13
制造技术与机床(2017年6期)2018-01-19
分析化学(2017年12期)2017-12-25
