
多层石墨烯的粗粒化分子动力学模型与力学性能
2016-12-22 07:32杨庆生尚军军
北京工业大学学报 2016年12期
杨庆生, 尚军军, 刘 夏
(北京工业大学机械工程与应用电子技术学院, 北京 100124)
多层石墨烯的粗粒化分子动力学模型与力学性能
杨庆生, 尚军军, 刘 夏
(北京工业大学机械工程与应用电子技术学院, 北京 100124)
为了有效研究多层石墨烯等石墨烯集合体的力学性能,避免由于实验难度大、全原子计算成本高等带来的困难,基于Tersoff势函数提出一种可用于计算大规模石墨烯的粗粒化分子动力学方法. 该方法计算时只需输入原子坐标,大大减少了模型的数据处理,提高了计算效率. 通过与全原子模型计算结果比较,本文提出的粗粒化分子动力学方法可以准确预测多层石墨烯的拉伸破坏性能,对制备高性能石墨烯纤维有一定的指导意义.
多层石墨烯;粗粒化模型;分子动力学模拟;力学性能;破坏强度
石墨烯是由单层碳原子通过sp2共价键结合而成的二维六边形网格结构,其特殊的结构使其具有优异的力学、电学、热学、化学等特性[1-3],是已知的最薄、最坚硬、最有韧性的新型材料,具有广阔的应用前景. 近年来,大规模石墨烯的成功制备使得石墨烯增强复合材料得到了很好的发展[4-5]. 石墨烯纤维[6]、石墨烯纸[7]、石墨烯气凝胶[8]等石墨烯集合体的问世进一步展示了石墨烯在纳米科技领域的应用潜力. 而石墨烯的尺寸、排布方式等对石墨烯集合体[9]及石墨烯复合材料[10]的性能有很大影响,因此充分掌握石墨烯材料的宏观变形与微观结构演变之间的关联具有重要意义. 由于尺度限制,实验方法在观测材料的纳米尺度演化规律方面具有很大的局限性,数值模拟方法成为重要的研究手段,分子动力学是主要研究方法之一. 大规模石墨烯及其多层级结构,如多层石墨烯等,具有跨尺度特性,全原子模拟费时费力. 粗粒化分子动力学在模拟介观尺度结构的同时又保留了系统的分子特性,是一种有效的多尺度模拟方法,被广泛应用于跨尺度纳米材料的研究. 如Liu等[11]采用粗粒化分子动力学方法深入研究了碳纳米管(carbon nanotube,CNT)连续纤维在加捻和拉伸条件下的力学行为及微观结构演变机理. 分子粗粒化模型也会受到材料结构的影响,Liu等[12]还根据CNT纤维的组成结构发展了一种粗粒化分子动力学模型研究其拉伸性能. 很多学者采用粗粒化分子动力学方法研究有关石墨烯的问题. Titov等[13]用粗粒化分子动力学方法研究了石墨烯在脂膜中的夹层结构;Wu等[14]模拟了烷基- 聚氧化乙烯在石墨烯表面的吸附及自组装行为;Pandey等[15]研究了肽与石墨烯片结合的稳定性;Cranford等[16]模拟了单层和多层石墨烯的自折叠行为;Cranford等[17]研究了加捻超长多层石墨烯带的结构和构象行为;Li等[18]研究了石墨烯与脂双层膜作用的机理;Wang等[19]重点研究了石墨烯厚度、氧化石墨烯、脂质分子表面吸附性对石墨烯与脂双层膜相互作用的影响. 目前,石墨烯的粗粒化分子动力学模型主要有2类[20],一类是Martini力场模型[21],一类是基于应变能守恒的降阶模型[16],由于这些方法在计算大变形问题时误差较大且不能反映石墨烯的层间剪切响应,因此不适用于模拟多层石墨烯的力学行为. Ruiz等[20]发展了一种可模拟石墨烯多层级结构大变形及破坏的粗粒化分子动力学方法. 该方法的系统势能由键能、键角能、二面角能及非成键势能构成,根据应变能守恒原理对各个参数进行校验,得到粗粒化分子动力学函数,其中键能函数形式为Morse势. 虽然该方法可以很好地模拟多层石墨烯的力学性能,但是由于其势能包含键角能和二面角能,在建模时需要模型的角度参数和二面角参数,因此当模型复杂时,建模比较困难. 本文基于Tersoff势函数,发展了一种可以对大规模石墨烯进行数值模拟的粗粒化分子动力学方法,模型输入数据简单,对多层石墨烯的拉伸破坏进行了有效预测.
1 粗粒化模型与参数
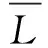
本文以Tersoff势函数形式表示粗粒之间的成键作用,Tersoff势函数的总能量函数为
(1)
Vij(r)=[aijfR(rij)+bijfA(rij)]fC(rij)
(2)
式中:fR为排斥势;fA为吸引势;fC为相互作用的截断函数;aij为阶段距离,当只考虑第一近邻距离时,aij≈1;bij为键序函数,用于表征系统的多体效应,它考虑了i、j原子及其周围键空间分布的影响[25-26].
Lernnard-Jones势用来描述同一分子体系内非化学键作用的两原子间的势能,表达式为
(3)
式中:r为两原子之间的距离;ε为能量标度参数;σ为碰撞直径参数.
对不同的n,势能函数中参数的取值也不相同,对于本文所研究的情况,即n=2,Tersoff势函数中发生变化的参数及变化后的取值如表1所示. Lernnard-Jones势函数的参数取值为ε=0.022 92 eV,σ=0.346 9 nm.

表1 粗粒化模型的参数
2 数值模拟
本文所有的分子动力学计算过程采用LAMMPS程序[27]执行. 为了验证粗粒化模型的准确性,对单层、双层和三层的石墨烯分别进行了全原子模型和粗粒化模型的拉伸模拟,并对结果进行了比较. 模拟中单片石墨烯的尺寸为11.912 nm×20.277 nm,其中x和y方向分别沿着石墨烯的zigzag和armchair方向. 该尺寸石墨烯对应的全原子模型中原子的个数为9 408,粗粒化模型中粗粒的个数为2 352. 双层石墨烯和三层石墨烯分别由2层和3层单片石墨烯堆叠而成,石墨烯片的层间距离为0.335 nm. 系统经过能量最小化后,采用NVE微正则系综,在300 K温度下对模型进行充分弛豫,时间步长设置为1 fs. 模型的底端固定,沿y方向拉伸,拉伸过程中温度保持不变,直到模型发生破坏.
本文用粗粒化分子动力学方法对多层石墨烯的拉伸性能进行了模拟,研究了石墨烯片不同排布方式对整体性能的影响. 为此选取4种排布方式的模型进行模拟,如图2所示,每个模型层数均为11,层间距离为0.335 nm,所有模型的整体尺寸均为45.928 nm×7.374 nm×3.685 nm. 当一层中有多片石墨烯时,相邻2片之间的距离为0.984 nm. A模型中,每层石墨烯都是完整的,模型中粗粒个数为37 224. B模型的石墨烯片有规律的排布,1,3,…,11层的石墨烯排布完全相同,每层有2片尺寸相同的石墨烯;2,4,…,10层的石墨烯排布完全相同,每层有3片尺寸相同的石墨烯,模型中总的粗粒个数为36 360. C模型中的石墨烯片随机分布. 为了与B模型具有可比性,C模型粗粒个数与B模型粗粒个数相同,每层均由2或3片石墨烯构成,石墨烯片的尺寸随机. 为了研究连续石墨烯片对多层石墨烯性能的影响,将C模型中的2、6、11层石墨烯替换为一整片完整石墨烯,其余层数石墨烯的组成方式不变,形成D模型,模型中粗粒个数为36 576,如图2(d)所示. 模拟过程如下:系统先经过能量最小化后,在NVE微正则系综中进行充分弛豫,弛豫温度控制在300 K,时间步长为1 fs. 系统得到最优化初始构型后沿y方向拉伸,直到发生断裂破坏.
3 结果与讨论
3.1 粗粒化模型准确性验证
由图3可以看出,对于单层石墨烯拉伸,粗粒化模型与全原子模型结果吻合得很好,单片石墨烯的弹性模量为0.94 TPa,与实验[28]和数值模拟[29]结果一致. 单片石墨烯的破坏强度为177 GPa,比实验测得数值(130±10)GPa[28]偏高,这可能是由于数值模拟模型为理想化材料所引起的. 图4所示为全原子模型和粗粒化模型拉伸破坏过程,二者破坏位置都是从底部固定端开始,裂纹扩展方向与轴线大致呈45°.
图5为双层石墨烯全原子模型和粗粒化模型的结果比较. 从势能和应力与拉伸应变的变化关系可以看出,全原子模型在第1层石墨烯发生断裂后,第2层石墨烯又被拉伸一段位移后断裂,2层分别断裂区分明显. 而对于粗粒化模型,2层石墨烯断裂时间间隔很短,模型在一层发生破坏后,整体迅速破坏,破坏模式如图6所示. 虽然2种模型在破坏过程中裂纹扩展的形式有所不同,但整体的破坏应变和破坏强度非常相近. 由于模型发生破坏后材料已经失效,而破坏之前2种模型的力学性能基本一致,因此可以用粗粒化模型来代替全原子模型.
三层石墨烯的全原子模型和粗粒化模型拉伸破坏的结果与双层石墨烯类似,2种模型的破坏强度和破坏应变相差很小,全原子模型不同层的破坏相隔时间较长,而粗粒化模型各层几乎同时断裂,如图7所示. 具体每层的破坏模式如图8所示.
3.2 不同排布方式的多层石墨烯的数值模拟结果
不同排布方式的多层石墨烯力学性能差异较大,因此图2所示模型的数值结果将在不同的图中表示. A模型的计算结果如图9所示. 由图9(b)可以看出,当多层石墨烯每层都由完整石墨烯片构成时,其破坏强度与单层石墨烯相差不大,但是破坏应变明显增加. 图10所示为B和C模型的计算结果. 从图10(b)中可以看出,B和C模型的破坏强度远低于A模型,这是因为B、C模型中,每层都是多片石墨烯组成,主要靠层间的范德华力来承受拉力,不同层的石墨烯片发生相对滑移引起整个结构失效,而单个石墨烯片并没有发生破坏. 由于B模型中石墨烯片发生相对滑移时,相互作用的表面积大于C模型,因此B模型的破坏强度要大于C模型. 为了研究石墨烯片中的成键作用和非成键作用对拉伸性能的共同影响,对D模型进行了模拟,结果如图11所示. 由结果可以看出,D模型的破坏强度比B、C模型明显增强.
4 结论
1) 本文提出的粗粒化分子动力学方法可以准确预测大规模石墨烯的拉伸性能.
2) 当多层石墨烯中每层的石墨烯片都不连续时,材料整体的破坏强度大大降低. 反之,含有连续石墨烯片的多层石墨烯具有较高的破坏强度.
[1] NOVOSELOV K S, GEIM A K, MOROZOV S V, et al. Electric field effect in atomically thin carbon films[J]. Science, 2004, 306(5696): 666-669.
[2] GEIM A K, NOVOSELOV K S. The rise of graphene[J]. Nature Materials, 2007, 6(3): 183-191.
[3] GEIM A K. Graphene: status and prospects[J]. Science, 2009, 324(5934): 1530-1534.
[4] ZHAO X, ZHANG Q, CHEN D, et al. Enhanced mechanical properties of graphene-based poly(vinyl alcohol) composites[J]. Macromolecules, 2010, 43(5): 2357-2363.
[5] KUILLA T, BHADRA S, YAO D, et al. Recent advances in graphene based polymer composites[J]. Progress in Polymer Science, 2010, 35(11): 1350-1375.
[6] XU Z, GAO C. Graphene chiral liquid crystals and macroscopic assembled fibres[J]. Nature Communications, 2011, 2: 571.
[7] CHEN H, MÜLLER M B, GILMORE K J, et al. Mechanically strong, electrically conductive, and biocompatible graphene paper[J]. Advanced Materials, 2008, 20(18): 3557-3561.
[8] ZHANG X, SUI Z, XU B, et al. Mechanically strong and highly conductive graphene aerogel and its use as electrodes for electrochemical power sources[J]. Journal of Materials Chemistry, 2011, 21(18): 6494.
[9] CONG H P, CHEN J F, YU S H. Graphene-based macroscopic assemblies and architectures: an emerging material system[J]. Chemical Society Reviews, 2014, 43(21): 7295-7325.
[10] ZHANG M, LI Y, SU Z, et al. Recent advances in the synthesis and applications of graphene-polymer nanocomposites[J]. Polymer Chemistry, 2015, 6(34): 6107-6124.
[11] LIU X, LU W, AYALA O M, et al. Microstructural evolution of carbon nanotube fibers: deformation and strength mechanism[J]. Nanoscale, 2013, 5(5): 2002.
[12] LIU X, YANG Q. Plastic deformation and failure mechanisms of collapsed-carbon nanotube fibers by coarse-grained molecular dynamic simulations[J]. International Journal of Plasticity, 2015, 64: 104-112.
[13] TITOV A V, KRL P, PEARSON R. Sandwiched graphene-membrane superstructures[J]. ACS Nano, 2010, 4(1): 229-234.
[14] WU D, YANG X. Coarse-grained molecular simulation of self-assembly for nonionic surfactants on graphene nanostructures[J]. The Journal of Physical Chemistry B, 2012, 116(39): 12048-12056.
[15] PANDEY R B, KUANG Z, FARMER B L, et al. Stability of peptide (P1 and P2) binding to a graphene sheet via an all-atom to all-residue coarse-grained approach[J]. Soft Matter, 2012, 8(35): 9101.
[16] CRANFORD S, SEN D, BUEHLER M J. Meso-origami: folding multilayer graphene sheets[J]. Applied Physics Letters, 2009, 95(12): 123121.
[17] CRANFORD S, BUEHLER M J. Twisted and coiled ultralong multilayer graphene ribbons[J]. Modelling and Simulation in Materials Science and Engineering, 2011, 19(5): 785-791
[18] LI Y, YUAN H, VON DEM BUSSCHE A, et al. Graphene microsheets enter cells through spontaneous membrane penetration at edge asperities and corner sites[J]. Proceedings of the National Academy of Sciences, 2013, 110(30): 12295-12300.
[19] WANG J, WEI Y, SHI X, et al. Cellular entry of graphene nanosheets: the role of thickness, oxidation and surface adsorption[J]. RSC Advances, 2013, 3(36): 15776-15782.
[20] RUIZ L, XIA W, MENG Z, et al. A coarse-grained model for the mechanical behavior of multi-layer graphene[J]. Carbon, 2015, 82: 103-115.
[21] MARRINK S J, RISSELADA H J, YEFIMOV S, et al. The MARTINI force field: coarse grained model for biomolecular simulations[J]. The Journal of Physical Chemistry B, 2007, 111(27): 7812-7824.
[22] MONTICELLI L, KANDASAMY S K, PERIOLE X, et al. The MARTINI coarse-grained force field: extension to proteins[J]. Journal of Chemical Theory and Computation, 2008, 4(5): 819-834.
[23] HSU D D, XIA W, ARTURO S G, et al. Systematic method for thermomechanically consistent coarse-graining: a universal model for methacrylate-based polymers[J]. Journal of Chemical Theory and Computation, 2014, 10(6): 2514-2527.
[24] XIA W, MISHRA S, KETEN S. Substrate vs. free surface: competing effects on the glass transition of polymer thin films[J]. Polymer, 2013, 54(21): 5942-5951.
[25] TERSOFF J. Modeling solid-state chemistry-interatomic potentials for multicomponent systems[J]. Physical Review B, 1989, 39(8): 5566-5568.
[26] TERSOFF J. Erratum: modeling solid-state chemistry: interatomic potentials for multicomponent systems[J]. Physical Review B, 1990, 41(5): 3248.
[27] PLIMPTON S. Fast parallel algorithms for short-range molecular-dynamics[J]. Journal of Computational Physics, 1995, 117(1): 1-19.
[28] LEE C, WEI X, KYSAR J W, et al. Measurement of the elastic properties and intrinsic strength of monolayer graphene[J]. Science, 2008, 321(5887): 385-388.
[29] JIANG J, WANG J, LI B. Young’s modulus of graphene: a molecular dynamics study[J]. Physical Review B, 2009, 80(11): 113405.
(责任编辑 吕小红)
Coarse-grained Molecular Dynamics Model and Mechanical Properties of Multi-layer Graphene
YANG Qingsheng, SHANG Junjun, LIU Xia
(College of Mechanical Engineering and Applied Electronics Technology, Beijing University of Technology,Beijing 100124, China)
To investigate the behavior of multi-layer graphene (MLG) and other graphene assemblies effectively, a coarse-grained molecular dynamics (CG-MD) method was developed, which conquers the limitations of experimental observations and full atom simulation to some extent. The CG-MD method based on Tersoff potential was used to simulate large scale graphene. The coordinates of the model were needed in the process of computation, which greatly reduced the time of modeling. The comparison between the results of full atom model and CG-MD model was made. It is proved that the CG-MD model can predict the tensile behavior of MLG accurately and it has great significance to producing graphene fiber with high performance.
multi-layer graphene;coarse-grained model; molecular dynamics simulation;mechanical properties;failure strength
2016- 08- 08
国家自然科学基金资助项目(11472020)
杨庆生(1962—), 男, 教授, 博士生导师, 主要从事新型材料与结构的力学问题方面的研究, E-mail: qsyang@bjut.edu.cn
TB 303
A
0254-0037(2016)12-1798-07
10.11936/bjutxb2016080021
猜你喜欢
昆钢科技(2022年1期)2022-04-19
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
小星星·阅读100分(低年级)(2021年8期)2021-08-27
纺织科学研究(2021年7期)2021-08-14
读者·校园版(2020年24期)2020-11-30
军事文摘(2020年20期)2020-11-16
小猕猴智力画刊(2013年1期)2013-03-15
学苑创造·B版(2009年10期)2009-11-16
