
参附益肾胶囊的质量标准研究
2016-12-15 06:13盛春帅李海霖青海省中医院西宁810000
中国药房 2016年33期
盛春帅,周 鹃,李海霖(青海省中医院,西宁 810000)
参附益肾胶囊的质量标准研究
盛春帅*,周 鹃,李海霖#(青海省中医院,西宁 810000)
目的:建立参附益肾胶囊的质量标准。方法:检测制剂中手参的显微特征;采用薄层色谱法(TLC)对制剂中丹参、大黄、甘草、红景天、肉桂进行定性鉴别;采用高效液相色谱法测定制剂中黄芪甲苷的含量:色谱柱为Phenomenex Luna-C18,流动相为乙腈-水(35∶65,V/V),流速为1.0 ml/min,柱温为30℃。结果:制剂中手参显微特征显著。丹参、大黄、甘草、红景天、肉桂TLC图斑点清晰,分离度好。黄芪甲苷检测进样量线性范围为0.832 8~4.164 0µg(r=0.999 9);精密度、稳定性、重复性试验的RSD≤2.03%;加样回收率为95.02%~104.33%(RSD=3.80%,n=6)。结论:该研究所建标准可用于参附益肾胶囊的质量控制。
参附益肾胶囊;质量标准;薄层色谱法;高效液相色谱法;黄芪甲苷
糖尿病肾病是糖尿病引起的危害性极大的一种慢性并发症。近些年,随着来我院内分泌、肾病专科就诊患者的不断增加,我院专家通过多年的临床观察,发现由于高海拔地区,自然氧气稀薄,清气不足,加之寒盛易伤阳气、干燥多风易伤阴津,使该地区糖尿病肾病多以阳虚血瘀型为主,故采用黄芪、午参、水蛭、大黄、附子等中、藏药组方,且在临床治疗糖尿病肾病、慢性肾炎、慢性肾功能不全中取得了很好的效果。为方便患者并配合临床治疗,医院将此组方制成胶囊作为医院制剂供临床使用。为有效控制该制剂质量,笔者对该制剂中的藏药手参进行了显微鉴别;对丹参、大黄、甘草、红景天、肉桂进行了薄层色谱法(TLC)鉴别;对制剂中主要成分黄芪甲苷采用高效液相色谱法(HPLC)进行了含量测定[1-4]。
1 材料
1.1 仪器
1260型HPLC仪,包括Ⅱ蒸发光散射检测器、四元泵(美国Agilent公司);BX51型显微镜(日本Olympus公司);BT224S型电子天平、XS105DU型电子天平(德国Sartrious公司);KQ5200B型超声波清洗器(昆山市超声仪器有限公司,功率:250 W,频率:50 kHz)。
1.2 药品与试剂
参附益肾胶囊(青海省中医院自制,批号:20140822、20141202、20140925,规格:0.4 g/粒);丹酚酸B对照品(批号:111562-201212,纯度>98%)、大黄素对照品(批号:110756-201319,纯度>98%)、红景天苷对照品(批号110818-201206,纯度>98%)、肉桂酸对照品(批号:110786-200503,纯度>98%)、黄芪甲苷对照品(批号:110831-200302,纯度>98%)、大黄对照药材(批号:120902-201010)、甘草对照药材(批号:120904-201318)、红景天对照药材(批号:121412-200902)均购自中国食品药品检定研究院;硅胶G薄层板(青岛海洋化工厂);甲醇、乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
1.3 药材
手参、红景天药材,购自青海省九康药材饮片有限公司,由本院赵克宏副主任药师鉴定为真品。
2 方法与结果
2.1 显微鉴别[5-7]
以处方中原粉药材入药的手参为对照,取本品适量,置显微镜下观察,可见草酸钙针晶束长24~50 μm,散在或存在于类圆形或椭圆形黏液细胞中,详见图1。
2.2 定性鉴别
2.2.1 丹参 取本品内容物3 g,研细,加70%乙醇30 ml,加热回流1 h,放冷,滤过,取滤液蒸干,残渣加水10 ml使溶解,用稀盐酸调pH至2,用乙酸乙酯振摇提取2次,每次20 ml,合并乙酸乙酯提取液,蒸干,残渣加乙醇1 ml使溶解,作为供试品溶液。另取丹酚酸B对照品,加70%乙醇制成丹酚酸B质量浓度为1 mg/ml的对照品溶液。按参附益肾胶囊处方和工艺制备缺丹参的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][8]试验,吸取上述3种溶液各10 μl,分别点于同一硅胶G薄层板上,以甲苯-三氯甲烷-乙酸乙酯-甲醇-甲酸(2∶3∶4∶0.5∶2,V/V/V/V/V)为展开剂,展开,取出,晾干,喷以3%三氯化铁乙醇溶液,置日光灯下检视。结果,供试品色谱中,在与对照品色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图2。

图1 手参的显微图Fig 1 Microscopic characteristics of Gymnadenia conopsea

图2 丹参的薄层色谱图1~3.供试品;4.对照品;5.阴性对照Fig 2 TLC chromatograms of Salvia miltiorrhiza1-3.test samples;4.reference substance;5.negative control
2.2.2 大黄 取本品内容物2 g,研细,加甲醇20 ml,超声处理20 min,滤过,取滤液蒸干,残渣加水10 ml使溶解,再加盐酸1 ml,置水浴上加热回流30 min,立即冷却,用乙醚振摇提取2次,每次20 ml,合并乙醚提取液,蒸干,残渣加甲醇1 ml使溶解,作为供试品溶液。另取大黄素对照品,加甲醇制成大黄素质量浓度为1 mg/ml的对照品溶液。再取大黄对照药材0.2 g,同供试品溶液制备方法制成对照药材溶液。按参附益肾胶囊处方和工艺制备缺大黄的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][8]试验,吸取上述4种溶液各5 μl,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1,V/V/V)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图3。
2.2.3 甘草 取本品内容物5 g,研细,加甲醇20 ml,超声处理30 min,滤过,滤液蒸干,残渣加水20 ml溶解,用水饱和的正丁醇提取2次,每次20 ml,合并正丁醇溶液,蒸干,残渣加甲醇2 ml使溶解,作为供试品溶液。另取甘草对照药材1 g,同供试品溶液制备方法制成对照药材溶液。按参附益肾胶囊处方和工艺制备缺甘草的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][8]试验,吸取上述3种溶液各10 μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-冰乙酸-水(15∶1∶1∶2,V/V/V/V)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇,于105℃下加热至斑点显色清晰,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图4。

图3 大黄的薄层色谱图1~3.供试品;4.对照药材;5.对照品;6.阴性对照Fig 3 TLC chromatograms of Rheum palmatum1-3.test samples;4.control medicinal herb;5.reference substance;6.negative control

图4 甘草的薄层色谱图1~3.供试品;4.对照药材;5.阴性对照Fig 4 TLC chromatograms of Glycyrrhiza uralensis1-3.test samples;4.control medicinal herb;5.negative control
2.2.4 红景天 取本品内容物5 g,研细,加甲醇20 ml,超声处理30 min,滤过,滤液蒸干,残渣加甲醇2 ml使溶解,作为供试品溶液。另取红景天苷对照品,加甲醇制成红景天苷质量浓度为1 mg/ml的对照品溶液。再取红景天对照药材5 g,同供试品溶液制备方法制成对照药材溶液。按参附益肾胶囊处方和制备工艺制备缺红景天的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][8]试验,吸取上述4种溶液各10 μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-丙酮-水(6∶3∶1∶1,V/V/V/V)的下层溶液为展开剂,展开,取出,晾干,置碘蒸气中熏约3 min,置日光灯下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图5。
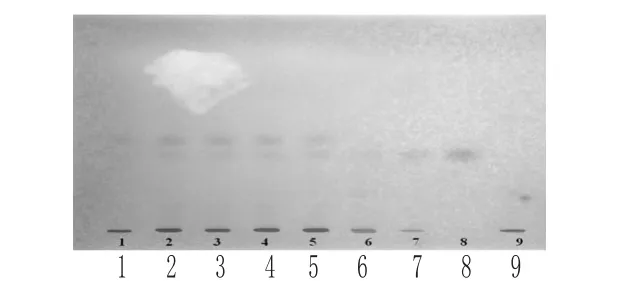
图5 红景天的薄层色谱图1~5.供试品;6.对照药材;7~8.对照品;9.阴性对照Fig 6 TLC chromatograms of Rhodiola rosea1-5.test samples;6.control medicinal herb;7-8.reference substances;9. negative control
2.2.5 肉桂 取本品内容物5 g,研细,加乙酸乙酯40 ml,超声处理20 min,滤过,滤液低温挥干,残渣加无水乙醇1 ml使溶解,作为供试品溶液。另取肉桂酸对照品,加无水乙醇制成肉桂酸质量浓度为1 mg/ml的对照品溶液。按参附益肾胶囊处方和工艺制备缺肉桂的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][8]试验,吸取上述3种溶液各10 μl,分别点于同一硅胶G薄层板上,以正己烷-乙醚-冰乙酸(5∶5∶0.1,V/V/V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照品色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图6。

图6 肉桂的的薄层色谱图1~3.供试品;4.对照品;5.阴性对照Fig 6 TLC chromatograms of Cortex cinnamomi1-3.test samples;4.reference substance;5.negative control
2.3 含量测定
2.3.1 色谱条件与系统适用性试验 色谱柱:Phenomenex Luna-C18(250 mm×4.6 mm,5µm);流动相:乙腈-水(35∶65,V/V);流速:1.0 ml/min;柱温:30℃。在上述色谱条件下,理论板数以黄芪甲苷峰计≥4 000;各成分基线分离良好,分离度>1.5;供试品溶液色谱图中,在与黄芪甲苷对照品色谱峰相应的位置上,有相同的色谱峰,阴性略有干扰,但从峰值上看,干扰极小,详见图7。
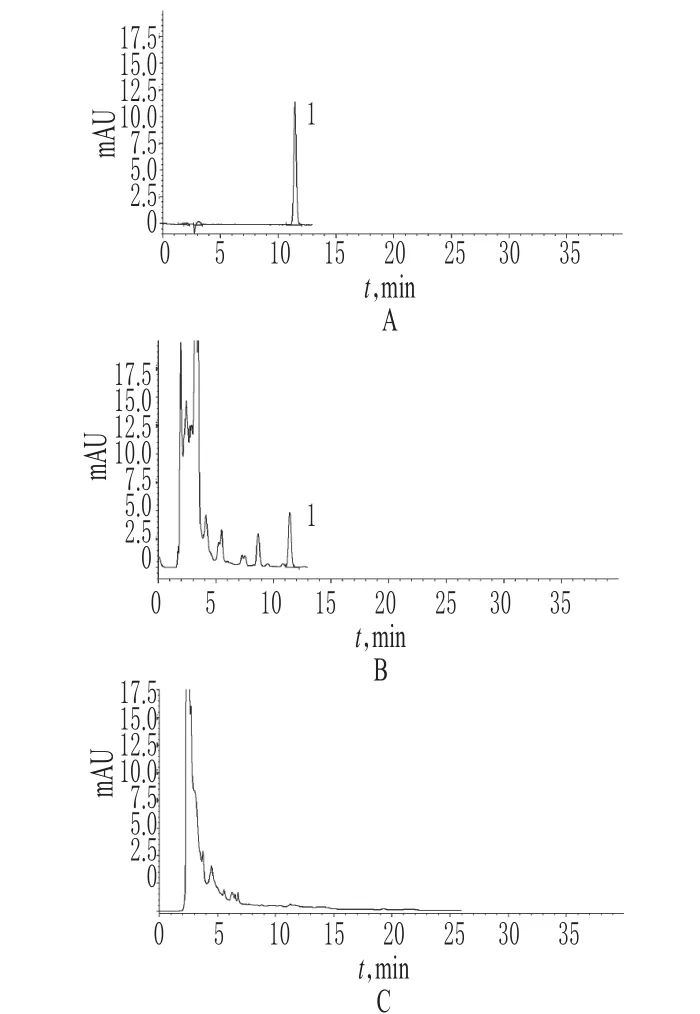
图7 高效液相色谱图A.对照品;B.供试品;C.阴性对照;1.黄芪甲苷Fig 7 HPLC chromatogramsA.reference substance;B.test sample;C.negative control;1.astragaloside
2.3.2 对照品溶液的制备 精密称取黄芪甲苷对照品0.010 41 g,置于50 ml量瓶中,加甲醇溶解并定容,摇匀,制成黄芪甲苷质量浓度为0.208 2 mg/ml的对照品溶液。
2.3.3 供试品溶液的制备 取本品内容物适量,研细,取10 g,精密称定,置于具塞锥形瓶中,加水50 ml,称定质量,超声处理40 min,放冷,称定质量,用水补足减失的质量,摇匀,滤过,精密量取续滤液25 ml,用水饱和的正丁醇振摇提取4次,每次20 ml,合并正丁醇提取液,用氨试液洗涤3次,每次30 ml,弃去,分取正丁醇液,蒸干,残渣用甲醇溶解并转移至5 ml量瓶中,加甲醇定容,摇匀,即得。
2.3.4 阴性对照溶液的制备 按参附益肾胶囊处方和工艺制备缺黄芪的阴性样品,并按“2.3.3”项下方法制成阴性对照溶液。
2.3.5 线性关系考察 分别精密量取“2.3.2”项下对照品溶液4、8、12、16、20µl,按“2.3.1”项下色谱条件进样测定,记录峰面积。以黄芪甲苷进样量(x,μg)为横坐标、峰面积(y)为纵坐标进行线性回归,得黄芪甲苷回归方程为y=3.564 9+1 281.1x(r=0.999 9)。结果表明,黄芪甲苷检测进样量线性范围为0.832 8~4.164 0µg。
2.3.6 精密度试验 取“2.3.2”项下对照品溶液适量,按“2.3.1”项下色谱条件连续进样测定6次,记录峰面积。结果,黄芪甲苷峰面积的RSD=0.30%(n=6),表明仪器精密度良好。
2.3.7 稳定性试验 取“2.3.3”项下供试品溶液(批号:20140925)适量,分别于室温下放置0、2、3、4、6、8 h时按“2.3.1”项下色谱条件进样测定,记录峰面积。结果,黄芪甲苷峰面积的RSD=0.53%(n=6),表明供试品溶液在8 h内基本稳定。
2.3.8 重复性试验 精密称取同一批样品(批号:20140925)适量,按“2.3.3”项下方法制备供试品溶液,共6份,再按“2.3.1”项下色谱条件进样测定,记录峰面积。结果,黄芪甲苷的平均含量为0.31 mg/g,RSD=2.03%(n=6),表明本方法重复性良好。
2.3.9 加样回收率试验 取已知含量样品(批号:20140925)适量,共6份,分别加入一定质量的黄芪甲苷对照品,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。
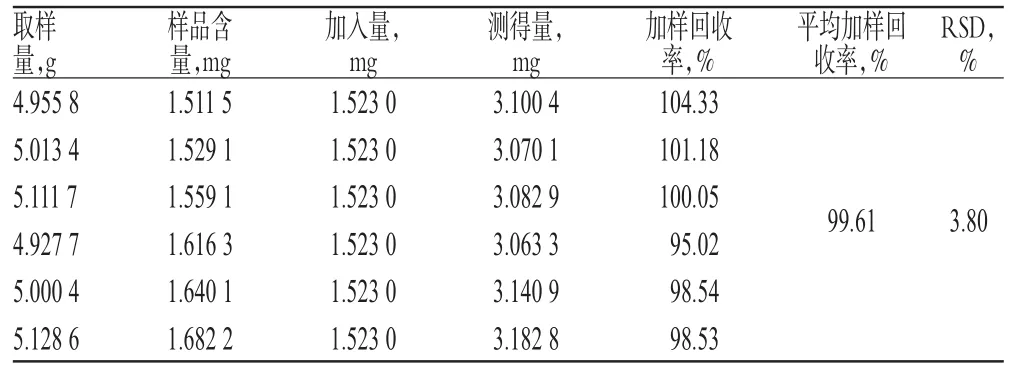
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery test(n=6)
2.3.10 样品含量测定 取3批样品适量,分别按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并计算样品含量,结果见表2。
3 讨论

表2 样品含量测定结果(n=3)Tab 2 Results of content determination(n=3)
本制剂为水提取的复方制剂,因而在TLC鉴别成分中以水溶性成分为对照品,除丹参素钠分离效果不好外,丹酚酸B、大黄素、红景天苷、肉桂酸,分离效果均较好,斑点清晰,因此将以上成分作为对照品列入标准。
考虑到黄芪提取方法的影响因素,笔者对提取过程中氨水洗涤因素、大孔吸附树脂因素、漂移管温度等进行了相关试验,最终确定了以氨水洗涤,选用D101型大孔吸附树脂柱,漂移管温度设定为50℃,这也使试验结果更加可靠。
从HPLC测定结果来看,由单方配方颗粒制成的样品含量略高于由药材提取制成的样品,这可能是药材混合提取时,各成分之间有相互抑制的作用,从而导致黄芪甲苷含量略低[9-10]。
综上所述,本研究所建标准可用于参附益肾胶囊的质量控制。
[1] 国家药典委员会.中华人民共和国药典:一部[S].2015年版.北京:中国医药科技出版社,2015:23、76、86、136、154.
[2] 国家药典委员会.中华人民共和国卫生部药品标准:藏药:第一册[S].北京:人民卫生出版社,1995:25、29、126、231.
[3] 国家食品药品监督管理局.国家食品药品监督管理局标准(试行)[S].YBZ12512005.
[4] 国家食品药品监督管理局.国家食品药品监督管理局标准(试行)[S].YBZ16142009.
[5] 青海省药品检验所,青海省藏医药研究所.中国藏药:第一卷[M].上海:上海科学技术出版社,1996:548.
[6] 青海省药品检验所,青海省藏医药研究所.中国藏药:第二卷[M].上海:上海科学技术出版社,1996:310.
[7] 徐国钧.中药材粉末显微鉴别[M].北京:人民卫生出版社,1986:52、416.
[8] 国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:57.
[9] 赵陆华.中药高效液相色谱法应用[M].北京:中国医药科技出版社,2005:326.
[10] 周训蓉,杨亮.金乌健骨胶囊的质量标准研究[J].中国药房,2015,26(30):4 247.
(编辑:张 静)
Study on Quality Standard of Shenfu Yishen Capsule
SHENG Chunshuai,ZHOU Juan,LI Hailin(Qinghai Province Hospital of Traditional Chinese Medicine,Xining 810000,China)
OBJECTIVE:To establish the quality standard for Shenfu yishen capsule.METHODS:The microscopic characteristics of Gymnadenia conopsea were detected;TLC was adopted for the qualitative identification of Salvia miltiorrhiza,Rheum palmatum,Glycyrrhiza uralensis,Rhodiola rosea and Cortex cinnamomi;HPLC was used for the content determination of astragaloside:the column was Phenomenex Luna-C18with mobile phase of acetonitrile-water(35∶65,V/V)at a flow rate of 1.0 ml/min,column temerpature was 30℃.RESULTS:Microscopic identification features of G.conopsea were exclusive,TLC identification of S.miltiorrhiza,R.palmatum,G.uralensis,R.rosea and C.cinnamomi was well separated and specific;the linear range of astragaloside was 0.832 8-4.164 0µg(r=0.999 9);RSDs of precision,stability and reproducibility tests were no higher than 2.03%;recovery was 95.02%-104.33%(RSD=3.80%,n=6).CONCLUSIONS:The standard can effectively control the quality of Shenfu yishen capsule.
Shenfu yishen capsule;Quality standard;TLC;HPLC;Astragaloside A
R284.1
A
1001-0408(2016)33-4735-04
2015-11-01
2016-03-16)
*主管药师。研究方向:中药制剂。E-mail:shengcs8587@163. com
#通信作者:主任药师。研究方向:中药制剂。E-mail:3345666989@qq.com
DOI 10.6039/j.issn.1001-0408.2016.33.43
猜你喜欢
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
海峡姐妹(2019年3期)2019-06-18
中成药(2018年9期)2018-10-09
中成药(2018年5期)2018-06-06
特别健康(2018年4期)2018-01-28
中成药(2017年3期)2017-05-17
中国药业(2014年12期)2014-06-06
中国药业(2014年24期)2014-05-26
中成药(2014年9期)2014-02-28
