
细胞自噬
2016-12-14 02:42:02Inductionofautophagyandinhibitionoftumorigenesisbybeclin
中国学术期刊文摘 2016年21期
关键词:癌症
Induction of autophagy and inhibition of tumorigenesis by beclin 1
Liang, XH; Jackson, S; Seaman, M; et al.
Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor
Yue, ZY; Jin, SK; Yang, CW; et al.
Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease
Ravikumar, B; Vacher, C; Berger, Z; et al.
Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice
Komatsu, M; Waguri, S; Ueno, T; et al.
细胞自噬调控的分子机制研究进展
李乐兴,戴汉川
细胞自噬的研究方法
马泰,孙国平,李家斌
细胞自噬
·编者按·
自噬(Autophagy,或称自体吞噬)是细胞将自身一些蛋白质和细胞器包裹在特定的膜结构中,送入溶酶体(酵母的液泡)降解,产生能量和小分子,如氨基酸等供细胞再次利用的过程。这是一个受到紧密调控的步骤,此步骤是细胞生长、发育与稳态中的常规步骤,它帮助细胞产物在合成、降解以及接下来的循环中保持一个平衡状态。细胞自噬的研究是目前生物医学领域热点之一,广泛参与各种生理和病理过程。日本科学家大隅良典因“对细胞自噬机制的发现”获得2016年度的诺贝尔生理学或医学奖。
1963年“自噬”的概念由比利时科学家Christian de Duve 在溶酶体国际会议上首先提出,Christian De Duve本人也因为开发了新的细胞研究手段,发现了溶酶体这一新的细胞器而获得1974年诺贝尔生理学或医学奖。当代的自噬研究是1990年代酵母的研究人员通过识别的自噬相关基因而被推动。
细胞自噬主要有3种形式:微自噬(microautophagy)、巨自噬(macroautophagy)和分子伴侣介导的自噬(Chaperone-mediated autophagy,CMA)。通常所说的自噬即指巨自噬,也是目前研究最多的。现在研究人员普遍采用电镜、免疫荧光、蛋白质印迹等方法检测自噬体及其标志蛋白。
本专题得到专家丁树哲教授(华东师范大学)的大力支持。
·热点数据排行·
截至 2016年10月19日,中国知网(CNKI)和Web of Science(WOS)的数据报告显示,以“细胞自噬”为词条可以检索到的期刊文献分别为 1035、 8569条,本专题将相关数据按照:研究机构发文数、作者发文数、期刊发文数、被引用频次进行排行,结果如下。

研究机构发文数量排名(CNKI)

研究机构发文数量排名(WOS)

作者发文数量排名(WOS)

(数据来源:中国知网、Web of Science,检索时间:2016-10-19)
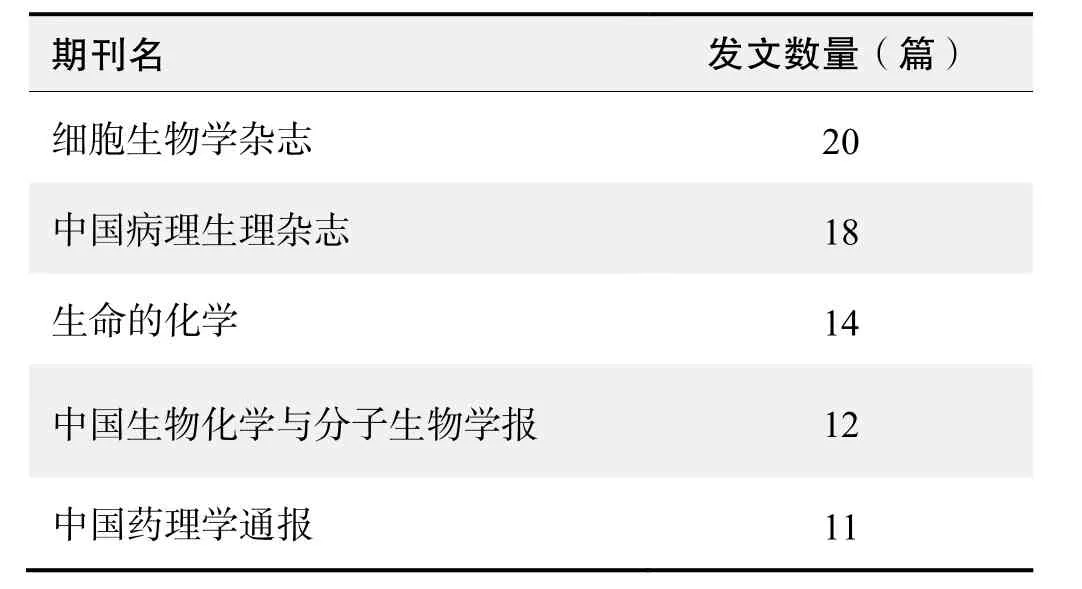
期刊发文数量排名(CNKI)

期刊发文数量排名(WOS)
根据中国知网(CNKI)数据报告,以“细胞自噬”等为词条可以检索到的高被引论文排行结果如下。

国内数据库高被引论文排行
根据Web of Science统计数据,以“细胞自噬”为词条可以检索到的高被引论文排行结果如下。

国外数据库高被引论文排行
·经典文献推荐·
基于Web of Science检索结果,利用Histcite软件选取LCS(Local Citation Score,本地引用次数)TOP 30文献作为节点进行分析,得到本领域推荐的经典文献如下。

本领域经典文献
来源出版物:FEBS Letters, 1993, 333(1-2): 169-174
Induction of autophagy and inhibition of tumorigenesis by beclin 1
Liang, XH; Jackson, S; Seaman, M; et al.
Abstract: The process of autophagy, or bulk degradation of cellular proteins through an autophagosomic-lysosomal pathway, is important in normal growth control and may be defective in tumour cells. However, little is known about the genetic mediators of autophagy in mammalian cells or their role in tumour development. The mammalian gene encoding Beclin 1, a novel Bcl-2-interacting, coiled-coil protein, has structural similarity to the yeast autophagy gene, apg6/vps30, and is mono-allelically deleted in 40%-75% of sporadic human breast cancers and ovarian cancers. Here we show, using gene-transfer techniques, that beclin 1 promotes autophagy in autophagy-defective yeast with a targeted disruption of agp6/vps30, and in human MCF7 breast carcinoma cells. The autophagy-promoting activity of beclin 1 in MCF7 cells is associated with inhibition of MCF7 cellular proliferation, in vitro clonigenicity and tumorigenesis in nude mice. Furthermore, endogenous Beclin 1 protein expression is frequently low in human breast epithelial carcinoma cell lines and tissue, but is expressed ubiquitously at high levels in normal breast epithelia. Thus, beclin 1 is a mammalian autophagy gene that can inhibit tumorigenesis and is expressed at decreased levels in human breast carcinoma. These findings
suggest that decreased expression of autophagy proteins may contribute to the development or progression of breast and other human malignancies.
来源出版物:Nature, 1999, 402(6762): 672-676
Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor
Yue, ZY; Jin, SK; Yang, CW; et al.
Abstract: The biochemical properties of beclin 1 suggest a role in two fundamentally important cell biological pathways: autophagy and apoptosis. We show here that beclin 1-/-mutant mice die early in embryogenesis and beclin 1+/-mutant mice suffer from a high incidence of spontaneous tumors. These tumors continue to express wild-type beclin 1 mRNA and protein, establishing that beclin 1 is a haploinsufficient tumor suppressor gene. Beclin 1-/-embryonic stem cells have a severely altered autophagic response, whereas their apoptotic response to serum withdrawal or UV light is normal. These results demonstrate that beclin 1 is a critical component of mammalian autophagy and establish a role for autophagy in tumor suppression. They both provide a biological explanation for recent evidence implicating beclin 1 in human cancer and suggest that mutations in other genes operating in this pathway may contribute to tumor formation through deregulation of autophagy.
来源出版物:Proceedings of the National Academy of Sciences, 2003, 100(25): 15077-15082
Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease
Ravikumar, B; Vacher, C; Berger, Z; et al.
Abstract: Huntington disease is one of nine inherited neurodegenerative disorders caused by a polyglutamine tract expansion. Expanded polyglutamine proteins accumulate abnormally in intracellular aggregates. Here we show that mammalian target of rapamycin (mTOR) is sequestered in polyglutamine aggregates in cell models, transgenic mice and human brains. Sequestration of mTOR impairs its kinase activity and induces autophagy, a key clearance pathway for mutant huntingtin fragments. This protects against polyglutamine toxicity, as the specific mTOR inhibitor rapamycin attenuates huntingtin accumulation and cell death in cell models of Huntington disease, and inhibition of autophagy has the converse effects. Furthermore, rapamycin protects against neurodegeneration in a fly model of Huntington disease, and the rapamycin analog CCI-779 improved performance on four different behavioral tasks and decreased aggregate formation in a mouse model of Huntington disease. Our data provide proof-of-principle for the potential of inducing autophagy to treat Huntington disease.
来源出版物:Nature Genetics, 2004, 36(6): 585-595
Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice
Komatsu, M; Waguri, S; Ueno, T; et al.
Abstract: Autophagy is a membrane-trafficking mechanism that delivers cytoplasmic constituents into the lysosome/vacuole for bulk protein degradation. This mechanism is involved in the preservation of nutrients under starvation condition as well as the normal turnover of cytoplasmic component. Aberrant autophagy has been reported in several neurodegenerative disorders, hepatitis, and myopathies. Here, we generated conditional knockout mice of Atg7, an essential gene for autophagy in yeast. Atg7 was essential for ATG conjugation systems and autophagosome formation, amino acid supply in neonates, and starvation-induced bulk degradation of proteins and organelles in mice. Furthermore, Atg7 deficiency led to multiple cellular abnormalities, such as appearance of concentric membranous structure and deformed mitochondria, and accumulation of ubiquitin-positive aggregates. Our results indicate the important role of autophagy in starvation response and the quality control of proteins and organelles in quiescent cells.
来源出版物:The Journal of Cell Biology, 2005, 169(3): 425-434
·推荐综述·
细胞自噬调控的分子机制研究进展
李乐兴,戴汉川
细胞自噬是广泛存在于真核细胞内的一种溶酶体依赖性的降解途径。细胞自噬导致细胞内长寿命蛋白和受损伤细胞器的降解,使细胞在应激条件下循环利用营养物质和三羧酸循环产生的ATP继续生存。根据细胞内底物运送到溶酶体腔方式的不同,哺乳动物细胞自噬可分为巨自噬(macroautophagy)、微自噬(microautophagy)和分子伴侣介导的自噬(chaperone-mediated autophagy,CMA)三种主要方式,但目前研究最为广泛的是巨自噬。研究表明,细胞自噬在细胞内稳态、癌症、心力衰竭、神经退行性疾病、传染病、衰老相关性疾病等生命过程中发挥着重要作用。近年来,随着自噬基因和及其功能被相继发现,自噬成为继凋亡(apoptosis)之后生命科学最热的研究领域之一。本文综述了近年来细胞自噬的研究进展,以期有助于细胞自噬调控的深入研究,并为治疗心脏疾病(如动脉粥样硬化)、癌症(如乳腺癌)等提供理论基础。
1 细胞自噬的过程
自噬包括生理条件的基础型自噬和应激条件下的诱导型自噬。细胞自噬主要包括以下过程:(1)自噬诱导,诸如辐射、饥饿、缺氧、细菌入侵、生长因子匮乏等多种因素均可诱导自噬发生;(2)囊泡(吞噬泡)成核,细胞内质网膜、线粒体外膜、高尔基复合体膜或质膜等构成囊泡的双层或多层膜结构,30多种 Atg(autophagy-related)蛋白在囊泡膜上顺序耦联组装成PAS(pre-autophagosomal structures)。其中,Beclin1/ BECN1(Atg6)作为构成III类PI3K(phosphoinositide 3-kinase)复合体的支架蛋白,通过Vps34-p150复合体与 Atg9、Atg14L 和 UVRAG(ultraviolet radiation resistance-associatedgene protein)等蛋白结合,形成PI3K核心复合体启动自噬;(3)自噬体的成熟,PI3K核心复合体转变成 PtdIns(3)P(phosphatidylinositol 3-phosphate),PtdIns(3)P作为“着陆台”,募集自噬蛋白进入到吞噬泡(phagophore)成核中心,在泛素化耦联系统的辅助下,Atg10、Atg7、Atg3、Atg8/LC3、 Atg4和Atg12-Atg5-Atg16L1等最终被募集到PAS,参与囊泡膜延伸和自噬体(autophagosome)的成熟;(4)自噬溶酶体的融合,自噬体通过酸化与溶酶体融合形成自噬溶酶体(autophagolysosome)。自噬体膜的受体蛋白SQSTM1/p62接受酶作用的底物(错误折叠蛋白或蛋白聚合体),降解 p62及其靶蛋白,清除线粒体、内质网等受损细胞器,释放出营养物质和ATP供细胞再利用。此外,p62还与自噬活性呈负相关性,反映自噬溶酶体溶解酶活性和自噬潮(autophagic flux)的强弱,因此可作为自噬的分子标记。
2 细胞自噬的功能
生理性自噬是细胞的自我保护机制,有益于细胞的生长发育,保护细胞防止代谢应激和氧化损伤,对维持细胞内稳态以及细胞产物的合成、降解和循环再利用具有重要作用;但自噬过度可能导致代谢应激、降解细胞成分、细胞死亡等,打破细胞生长和死亡(细胞死亡至少分为三种形态学上不同的进程,即细胞凋亡、自噬性细胞死亡和坏死,此处所指的死亡可能伴随着细胞自噬,过度自噬以一种不同于凋亡和坏死的方式使细胞死亡,但自噬与二者还有一定的关联性,比如 Bcl-2和Beclin1之间的互作)间的平衡。自噬在多种生理病理过程中发挥重要作用。缺血再灌注显著上调Beclin1,激发心肌细胞自噬;Beclin1表达下调抑制自噬,减弱心肌损伤。研究认为,自噬是动脉粥样硬化发展过程中的一种保护机制,因为自噬通过加工氧化修饰蛋白使斑块固化,而自噬缺陷加剧动脉粥样硬化。在肿瘤细胞生物学上,根据细胞基因组成和细胞所处环境的变化,自噬既能抑制肿瘤抑制因素发挥作用,自噬基因的缺失又可促进肿瘤的生成。研究发现,一方面,自噬可使乳腺癌细胞继续生存;而另一方面,自噬又可导致结肠癌细胞HCT116的死亡。目前普遍认为,在乏氧、营养缺乏、代谢应激等条件以及抗癌治疗(如化学疗法、放射疗法)等环境下,癌细胞通过自噬可以继续生存。此外,自噬还可能在衰老、炎症、细胞凋亡、胞内病原体入侵、神经退行性疾病等方面发挥着重要作用。
3 细胞自噬调控的分子机制
3.1 泛素样蛋白系统对细胞自噬的调控
泛素化是在翻译后水平上进行蛋白修饰的一种方式,参与蛋白酶体依赖性蛋白水解、蛋白功能调控、亚细胞分布和/或蛋白质互作。在泛素激活酶(ubiquitin-activating enzyme,El)、泛素接合酶(ubiquitin-conjugating enzyme,E2)以及泛素蛋白连接酶(ubiquitin-protein ligase,E3)的连续作用下,泛素与底物蛋白特定的Lys残基共价结合完成泛素化。同时,泛素化也是一种可逆性的过程,可由去泛素化酶将泛素从蛋白质上除去。泛素化主要包括以下3步酶促反应过程:(1)在ATP作用下,E1可在其Cys和泛素的C-端的Gly之间形成巯酯键,即E1-SH~Ub,从而激活泛素;(2)在ATP和E2酶作用下,泛素从E1转移到E2上,同样以巯酯键的方式结合(E2-SH~Ub);(3)E3酶可以特异性识别底物蛋白并与之结合,与此同时 E2将激活的泛素直接转移到某些 E3结合的底物上,经过多个重复,多个泛素之间通过Lys相互连接,在底物上形成多泛素链。E1-样酶Atg7和E2-样酶Atg10泛素样反应后,泛素样蛋白Atg12与Atg5 Lys130共价耦联,Atg16L1作为连接蛋白,增强Atg12和E3泛素连接酶样蛋白Atg5间的互作,而后Atg12-Atg5与Atg16L1形成E3连接酶样复合体并定位于PAS。半胱氨酸酶Atg4酶切LC3并暴露C-端最后5个Gly残基,在E2-样酶Atg3辅助下,与磷脂酰乙醇胺(phosphatidylethanolamine,PE)发生E3-样共轭形成脂化的LC3(LC3-II)并定位于PAS,吞噬泡加工成为成熟自噬体。
3.2 mTOR信号通路对细胞自噬的调控
mTOR(mammalian target of rapamycin)属于Ser/Thr激酶,参与细胞发育、核糖体生成和代谢调控等生物学过程。mTOR包括雷帕霉素敏感型mTORC1和雷帕霉素非敏感型 mTORC2。mTORC1通过磷酸化ULK1-Atg13-RB1CC1-C12orf44/Atg101复合体使其失活,从而负调控细胞自噬体的形成,其活化程度可反映自噬水平,如果阻断mTORC1的功能,Ser/Thr激酶可磷酸化Atg1复合体并激活自噬。mTORC2的磷酸化能激活 Akt(PKB)和 Atg1抑制自噬,也可上调 HIF1A(hypoxia-inducible factor 1A)的表达。mTOR调控细胞自噬主要包括mTOR非依赖性和mTOR依赖性两条信号通路。
3.2.1 mTOR非依赖性信号通路
有实验发现,Mst1(mammalian Ste20-like kinase 1)可使 Beclin1BH3结构域 N-端的 Thr108磷酸化,增强Beclin1与Bcl-2和/或Bcl-xL疏水沟α3螺旋间的互作,使Beclin1同源二聚体稳定,减弱Atg14L与Beclin1的结合,降低 Beclin1-PI3K-Atg14L复合体脂激酶 Vps34的活性以抑制自噬。Molejon等认为,VMP1(vacuole membraneprotein 1)20位氨基酸残基C-端亲水性结构域(VMP1-AtgD)与Beclin1 BH3结构域结合致使Bcl-2与Beclin1解离,最终形成VMP1-Beclin1-hVps34-Atg14L复合体共同定位于PAS,启动PI3P生成、泛素样级联反应和囊泡的形成。有趣的是,棉酚衍生物ApoG2与Mst1作用相反,ApoG2破坏Beclin1和Bcl-2/xL的互作,释放出 Beclin1 BH3结构域,从而诱导自噬,但氯喹(chloroquine,CQ)与 ApoG2结合可阻断自噬体与溶酶体的融合。而EGFR(epidermal growthfactor receptor)通过磷酸化Beclin1多个位点的酪氨酸,增强Beclin1与抑制剂的结合能力,降低Vps34脂激酶活性以抑制自噬。
3.2.2 mTOR依赖性信号通路
Qased等发现,Ser/Thr蛋白激酶 ATM(ataxia telangiectasia mutated)属 PIKK(PI3K-related protein kinase)家族,ATM C-端序列与PI3K催化区同源,其能够刺激 LBK/AMPK/TSC2通路的下游信号,抑制mTORC1。mTORC1被抑制后可激活 ULK1(unc-51 like autophagy activatingkinase 1),ULK1通过与UVRAG结合再使 Beclin1Ser14磷酸化,从而增强 Beclin1-Vps34-Atg14L复合体的活性,启动自噬。此外,作为ULK复合体的重要组成成分,FIP200的缺失会造成MEF(mouse embryonicfibroblast)的Atg14-Atg1-WIPI诱导缺陷。然而,Efeyan等指出,Rag GTPases活化后能够募集mTORC1进入到溶酶体表面导致自噬缺陷。除前述作用外,ApoG2亦可抑制线粒体电子传递以产生ROS(reactive oxygen species),ROS通过提高 ERK(extracellular regulated protein kinases)、JNK(c-Jun N-terminal kinases)(靶作用于Bim和Atg5)的磷酸化水平,加快HMGB1(high-mobility group box 1)从细胞核到细胞质的转运,以及抑制mTOR信号,启动细胞自噬。但NAC(N-acetyl-cysteine)可减弱HMGB1从细胞核到细胞质的转运,诱导细胞凋亡和杀伤。同样,EGFR也可使PI3K、Akt和mTOR的酪氨酸磷酸化,负调控细胞自噬。
3.2.3 其他信号对细胞自噬的调控
研究表明,在细胞核中,p53可通过sestrin1/2蛋白激活AMPKmTORC1信号通路,从而抑制mTORC1以
上调自噬水平;也可通过激活DAPK1(death-associated proteinkinase 1),磷酸化Beclin1,促进细胞自噬;还能通过激活抗凋亡蛋白 Bcl-2家族,解除 Bcl-2/xL与Beclin1之间的抑制作用而上调细胞自噬。而在细胞质中,p53缺失的癌细胞的自噬水平上调,重新载入 p53后可下调细胞自噬水平。还有研究表明,脂多糖(lipopolysaccharide,LPS)可通过 TLR(Toll like receptor)调节细胞自噬的水平。在天然免疫研究中发现,LPS能诱导小鼠单核巨噬细胞和人巨噬细胞自噬体形成,抑制TLR4后自噬体形成明显减少。LPS/TLR4信号通路介导的自噬可加强TLR4信号通路中髓样分化蛋白(myeloid differentiation factor 88,MyD88)或IFN诱导接头蛋白[Toll/interleukin (IL)-1receptor homology domain (TIR)-containing adaptorinducing interferon(IFN)-β,TRIF]与自噬蛋白 Beclin1的相互作用,抑制Beclin1和自噬信号通路中Bcl-2的结合,增强NF-κB核转录因子的活性。
此外,PI3K-Akt-FoxO信号通路可介导谷氨酰胺合成酶的活化,参与募集Atg蛋白,提高LC3和ULK2的共定位水平。ox-LDL(oxidized low-densitylipoprotein)极大地促进动脉粥样硬化的发生、发展,适当浓度(10~40 μg/mL)的ox-LDL可激活保护性细胞自噬,致使内皮细胞、血管细胞和巨噬细胞的溶酶体降解ox-LDL。
3.3 miRNA对细胞自噬的调控
microRNA(miRNA)是一类长约22 nt的内源性非编码小 RNA分子,在转录后水平调控基因的表达。研究表明,miRNA参与细胞生长发育、炎症、肿瘤、衰老、凋亡等多种生理病理过程。近年来,还发现miRNA参与了细胞自噬调控,在自噬的发生和形成中发挥重要作用。miRNA与其靶mRNA 3′-UTR部分互补序列配对,通过降解mRNA和/或抑制蛋白翻译来调控基因表达,并且miRNA与其靶mRNA的序列同源性决定了是降解mRNA还是抑制翻译。营养饥饿、缺氧、雷帕霉素等可诱导细胞自噬,但多数 miRNA在自噬过程的不同阶段可通过作用于Atg蛋白以拮抗这种诱导作用,抑制细胞自噬,对细胞造成伤害,且无细胞特异性。
3.3.1 囊泡(吞噬泡)成核阶段
在正常生长条件下,抗凋亡蛋白家族 Bcl-2(包括Bcl-2、Bcl-xL、Mcl-1、A1、Bcl-W和Rubicon)与Beclin1结合能力最强,Beclin1BH3结构域与Bcl-2和/或Bcl-xL的疏水沟互作,负调控Beclin1-Vps34 PI3K-p150核心复合体的形成和活性,形成Beclin1同源二聚体抑制自噬;当自噬被诱导时,Beclin1与Bcl-2解离启动自噬。此外,Beclin1 CCD(coiled-coil domain)结构域也可与Bcl-2和/或Bcl-xL的BH4结构域互作,但miR-376b、miR-216a、miR-30a、miR-30d都可靶向 Beclin1,抑制其表达,减弱Beclin1与Bcl-2的结合和解离能力,从而调控自噬。Atg9作为唯一的跨膜蛋白,定位于PAS、线粒体和高尔基复合体,启动脂质从生物膜转运到PAS,介导组装完整的囊泡膜,但miR-34a抑制Atg9A表达,中断囊泡成核。Atg14L可以调节脂激酶 Vps34活性,并可募集 ULK1以使 Beclin1磷酸化,但 miR-195靶抑制Atg14,以抑制细胞自噬。PI3KC3是PI3K复合体的核心蛋白,miR-338-5p通过抑制PI3KC3的表达,阻断囊泡成核,从而负调控细胞自噬。
3.3.2 自噬体的成熟阶段
miR-216a、miR-181-a、miR-30a和miR-30d靶作用于Atg5,miR-375、miR-199a-5p靶作用于Atg7,miR-23b、miR-30d靶作用于 Atg12,抑制泛素化蛋白表达,负调控囊泡膜延伸。除可脂化LC3-I外,Atg4酶(Atg4B)同样可以介导自噬体膜表面的LC3蛋白发生去脂化作用,从而使细胞自噬循环利用LC3蛋白。研究发现,miR-376b靶作用于Atg4C、miR-101靶作用于Atg4D以抑制细胞自噬。Suzuki等认为,LC3侧链lys49参与调控LC3和Atg13 C-端LIR(LC3 interactingregion,氨基酸序列H441D442D443F444V445M446I447)间的互作。miR-204通过靶向抑制LC3B的表达,阻断LC3B的脂化,从而抑制自噬体的成熟。LC3-II与自噬体形成有关,而未成熟的LC3-I是可溶的。因此,LC3-II或LC3-II/LC3-I比值常用作自噬体形成的分子标记,用于自噬现象的观察和分析。
3.3.3 自噬溶酶体的融合阶段
研究认为,Atg8不仅参与膜延伸,且可经接头蛋白LIR结构将错误折叠或聚合蛋白质、受损细胞器、病原体募集到自噬体膜进行降解。酵母 Atg8的哺乳动物同源蛋白有7种,即LC3A、LC3B、LC3C、GABARAP、GABARAPL-1、GABARAPL-3、GABARAPL-2,因此LC3亚型与Atg13 LIR亲和力的特异性也是影响自噬体形成的关键因素。但miR-204通过靶抑制LC3B表达,抑制自噬,从而降低自噬溶酶体的分解能力。
此外,miR-22、miR-138、miR-302b、miR-210分别靶作用于HMGB1、Mst1、EGFR、VMP1,且miR-22与HMGB1、Mst1与miR-138、miR-302b与EGFR、miR-
210与VMP1均呈负相关,这些miRNA通过抑制蛋白表达,从而负调控前述细胞自噬信号通路。Ucar等认为,IGF-1(insulinlike growth factor-1)可上调miR- 212/132的表达,并且miR-212/132靶抑制FoxO3表达以致弱细胞自噬。miR-132还可激活PI3K-Akt-mTOR信号通路,抑制细胞自噬。hsa-let-7g可能通过下调 LOX-1、ROS的表达,以抑制细胞自噬。miR-101还可靶作用于STMN1(在一定程度上拮抗 miR-101的抑制作用)、RAB5A,miR-30d靶作用于Atg2,miR-130a靶作用于Atg2B、DICER1,miR-224(可被自噬降解)可能靶作用于Smad4,这些均能够抑制细胞自噬。
研究还发现,除多数miRNA对细胞自噬有抑制作用外,少数miRNA也可增强细胞自噬水平(表2)。Wan等认为,miR-155、miR-7可靶作用于mTOR信号多种分子,如RHEB、RICTOR和RPS6KB2等,负调控PI3KAkt-mTOR信号通路,诱导细胞自噬。此外,miR-99a也可通过抑制mTOR信号、miR-18a通过上调ATM的表达以增强细胞自噬水平。
目前,细胞自噬与miRNA关系的研究还处于初级阶段,关于miRNA如何参与调控自噬的研究有待进一步阐明。随着研究的深入,不同的miRNA在细胞自噬中的作用与机制将会更加清楚,对理解机体病理生理过程、抗感染以及天然免疫机制等方面具有重要意义。在自噬过程涉及的复杂的分子调节网络中,解析 miRNA与细胞自噬关系,有助于寻找新的自噬调控靶点,也为揭示自噬分子调控机制提供新的思路和策略。
3.4 细胞自噬调控的其他机制
caspase属于半胱天冬氨酸蛋白酶(cysteinyl containing aspartate-specific protease),在正常细胞中,caspase以无活性的酶原形式存在。MOMP(mitochondrialouter membrane permeabilisation)是激活caspase的主要机制之一,细胞色素C进入到细胞质启动凋亡复合体的形成,激活 caspase-9,caspase-9被裂解后激活 caspase-3和caspase-7。细胞应激激活caspase,活化的caspase能够启动细胞降解,调控细胞凋亡。研究发现,ATP合成增加能够抑制细胞自噬,而凋亡蛋白caspase Dcp-1调控自噬潮(autophagicflux)的发生。pro-Dcp-1在线粒体聚集,Dcp-1与核苷酸转移酶SesB互作,降低SesB的稳定性并下调ATP的合成,从而启动细胞自噬。Atg16L1蛋白发生Thr300Ala/Thr316Ala突变,能够致使caspase-3对Atg16L1的降解敏感性增强,进一步影响自噬体的形成。但此时,36 kDa和34 kDa裂解产物的出现,并不是由于 caspase-3活性增强所造成的。calpain是一种钙依赖性的半胱氨酸蛋白酶,可在 Atg5蛋白的氨基末端进行裂解,产生一个相对分子质量为24000的产物,从而驱使细胞自噬向细胞凋亡转变,将自噬与凋亡联系起来。除Atg5蛋白外,Beclin1、Bax也是calpain的作用靶点,更说明自噬与凋亡有一定的关联性。
AMPK(AMP-activated protein kinase)是一种异源三聚体,包括α催化亚基1个和调控型β、γ亚基各1个。多种应激因素可激活AMPK,AMPK活化后既可抑制自噬,又可激活自噬。细胞渗透性核苷酸类似物AICAR(AICA riboside)可以激活肝细胞AMPK,AMPK磷酸化水平的提高能够抑制细胞自噬,致使神经元胞内泛素化蛋白的累积;而在酵母和多种哺乳动物细胞内,AMPK的活化可以激活自噬。Yu等认为,蛋白磷酸酶PP2A(protein phosphatase 2A)调控AMPK α和γ亚基间的互作,使AMPK α亚基去磷酸化,从而使AMPK抑制mTOR磷酸化,启动细胞自噬。
研究还认为,蛋白乙酰化是多种细胞进程的关键调控机制。Yi等对酿酒酵母进行遗传学分析证实,自噬过程必需组蛋白乙酰转移酶Esa1的参与,且Atg3是Esa1的作用底物。Atg3的 K19和 K48发生乙酰化,控制Atg3-Atg8互作和Atg8的脂化,进而调控细胞自噬。去除脱乙酰酶Rpd3后,提高K19-K48的乙酰化水平,可以增强细胞自噬。
4 结语与展望
在植物、酵母、蠕虫、鼠和人等真核生物中都发现细胞自噬现象,其与多种生理病理过程密切相关,成为生命科学领域研究的热点之一。虽然,目前的研究在一定程度上阐明了细胞自噬调控的分子机制,但极其复杂的自噬过程因位置、状态、环境、疾病的不同,信号通路、生物功能也会有所不同。因此,自噬体膜来源、自噬溶酶体融合机制、自噬相关蛋白功能、自噬调控网络以及尚存争议的自噬对细胞生存与死亡所发挥的双重作用等,仍待进一步研究,以期为人类预防和治疗肿瘤、免疫性疾病、神经退行性疾病和代谢性疾病等带来新的希望。♦
【作者单位:华中农业大学动物医学院】
(摘自《中国细胞生物学学报》2015年2期)
·高被引论文摘要·
被引频次:52
细胞自噬的研究方法
马泰,孙国平,李家斌
细胞自噬的研究是目前生物医学领域热点之一,广泛参与各种生理和病理过程。目前普遍采用的自噬检测方法包括电镜、免疫荧光、蛋白质印迹等方法检测自噬体及其标志蛋白。研究的深入对自噬的检测方法也提出了更高的要求,自噬功能障碍包括自噬体形成和降解障碍,因此,准确全面地评估自噬不仅包括自噬体的检测,还包括动态观察整个自噬性降解的过程是否顺畅(即自噬潮分析)。另外,通过药物或基因干预技术来人为地调控自噬以观察其在体内体外模型中的作用也是自噬分析的重要内容。需要注意的是,任何一种方法单独应用均不能作为自噬的依据,对任何方法得到的结果进行解释时必须慎重,特别是不能将自噬体的增多减少或自噬相关蛋白表达的高低等同于自噬的增强或减弱。
细胞自噬;自噬体;微管相关蛋白1轻链3;自噬潮;检测方法
来源出版物:生物化学与生物物理进展, 2012, 39(3): 204-209
被引频次:44
细胞自噬与肿瘤发生的关系
王宠,张萍,朱卫国
摘要:细胞自噬(autophagy)是将细胞内受损、变性或衰老的蛋白质以及细胞器运输到溶酶体进行消化降解的过程。正常生理情况下,细胞自噬利于细胞保持自稳状态;在发生应激时,细胞自噬防止有毒或致癌的损伤蛋白质和细胞器的累积,抑制细胞癌变;然而肿瘤一旦形成,细胞自噬为癌细胞提供更丰富的营养,促进肿瘤生长。因此,在肿瘤发生发展的过程中,细胞自噬的作用具有两面性。尽管大多数抑癌蛋白可以激活细胞自噬这一结论被广泛接受,但 p53作为重要的抑癌蛋白,在细胞核和细胞浆不同的亚细胞定位中对细胞自噬有着截然相反的调控。对于细胞自噬和癌症发生之间关系亟待深入的研究,这将会有助于人类更好地认识并最终攻克癌症。本文将针对细胞自噬与肿瘤发生过程中主要的信号调节通路展开介绍。
关键词:细胞自噬;癌症;Beclin1;FoxO;mTOR;p53
来源出版物:中国生物化学与分子生物学报, 2010, 26(11): 988-997
被引频次:41
细胞自噬与肿瘤
杜海磊,邱伟华,杨卫平
摘要:自噬又称为 II型程序性细胞死亡(typeII programmed cell death)是以胞质内出现双层膜结构包裹长寿命蛋白和细胞器的自噬体为特征的细胞“自我消化”的一系列生化过程。自噬现象最早是 Ashford和Porter于1962年用电子显微镜在人的肝细胞中观察到。近年来随着酵母模型的建立,分子生物学及基因技术的发展,对自噬的研究有了很大的进展。
关键词:自吞噬作用;细胞凋亡;溶酶体;肿瘤;信号转导
来源出版物:中国病理生理杂志, 2010, 26(2): 401-404
被引频次:39
冬凌草甲素通过诱导人宫颈癌HeLa细胞自噬下调凋亡的机制
崔侨,田代真一,小野寺敏,等
摘要:研究冬凌草甲素通过诱导人宫颈癌 HeLa细胞自噬拮抗凋亡的机制。MTT法测定冬凌草甲素对HeLa细胞的细胞毒作用。通过相差显微镜观察细胞形态学变化,用琼脂糖凝胶电泳检测DNA片段化,用流式细胞仪检测细胞自噬和凋亡水平,用Westernblotting检测分析药物对蛋白质表达的影响。冬凌草甲素明显抑制HeLa细胞的增殖,诱导HeLa细胞凋亡,同时诱导HeLa细胞发生自噬。Westernblotting检测结果表明,冬凌草甲素作用24 h后,促凋亡蛋白Bax、细胞色素c和控制Bax活力的去乙酰化酶SIRT-1的表达明显改变。冬凌草甲素(64 μmol·L-1)诱导的自噬通过影响SIRT-1和线粒体途径蛋白的表达下调凋亡。
关键词:冬凌草甲素;HeLa细胞;自噬;细胞凋亡
来源出版物:药学学报, 2007, 42(1): 35-39
被引频次:36
细胞自噬在肿瘤中作用的研究进展
李国东,吴德全,李本义
摘要:自噬(autophagy)是广泛存在于真核细胞中的基本生命现象,是程序性细胞死亡形式之一,近年来备受关注。自噬活性的变化与人类肿瘤的发生、发展密切相关,并可从多个层面影响肿瘤进程,包括肿瘤细胞凋亡、血管生成及抗肿瘤治疗等。本文就自噬及其在肿瘤中作用的研究进展进行简要综述。
关键词:自噬;肿瘤;信号调控;肿瘤治疗
来源出版物:癌症, 2009, 28(4): 445-448
被引频次:36
缺血/再灌注过程中心肌细胞自噬研究进展
李欣志,刘建勋
摘要:自噬是一种广泛存在于真核细胞中的生命现象,心肌细胞营养缺乏、缺血/再灌注损伤、心衰等均可诱发细胞自噬。缺血/再灌注过程中的心肌细胞自噬可以维持心肌细胞稳态、减少细胞缺失,但是自噬作用也可导致心肌细胞死亡。
关键词:自噬;缺血/再灌注;心肌细胞
来源出版物:中国药理学通报, 2008, 24(6): 704-707
被引频次:32
自噬的抑制影响长春新碱诱导的肝癌细胞自噬性凋亡
彭心昭,陈英,朴英杰
摘要:目的:研究自噬特异性抑制剂3-甲基腺嘌呤(3-methyladenine,3MA)对长春新碱(vincristine,VCR)诱导的肝癌细胞系 HepG2细胞自噬性凋亡的影响。方法:利用已建立的VCR诱导HepG2细胞自噬性凋亡之模型,采用monodansylcadaverin(MDC)染色及流式细胞术荧光强度测定方法对自噬进行定量研究,采用流式细胞术及DNA电泳检测细胞凋亡。结果:3MA可以特异性地阻止自噬泡形成,还可以部分地抑制HepG2细胞自噬性凋亡。结论:自噬是VCR诱导的HepG2细胞自噬性凋亡所必需的。
关键词:自吞噬作用/药物作用;凋亡;长春新碱;3-甲基腺嘌呤
来源出版物:肿瘤, 2004, 24(1): 32-34
被引频次:31
淋巴细胞自噬性凋亡的超微结构细胞化学研究
朴英杰,黄行许,霍霞,等
摘要:本实验用透射电镜和细胞化学染色法观察放线菌酮处理和γ射线照射大鼠胸腺、脾脏和肠系膜淋巴结的超微结构。结果显示,放线菌酮注射后4 h,或γ射线照的2 h后引起大鼠胸腺、脾脏及肠系膜淋巴结中的淋巴细胞凋亡。凋亡淋巴细胞发生一系列的形态学变化,其胞核染色质凝集、重排,使核呈不同的形状;凋亡淋巴细胞的线粒体、内质网、溶酶体等细胞器大量增殖,我们称之为细胞反跳现象;大部分凋亡淋巴细胞凋亡时呈现典型的自噬性凋亡特征,即内质网大量增殖、分割、包裹凋亡淋巴细胞内变性的细胞成分形成大量自噬体,自噬体和溶酶体融合成为自噬佐调亡小体,和非自噬性凋亡形成的凋亡小体一起,由巨噬细胞消化分解。
关键词:自噬性凋亡;淋巴细胞;放线菌酮;γ射线:超微细胞化学
来源出版物:第一军医大学学报, 1996, 16(3): 165-171
被引频次:25
自噬参与心脏疾病调控的研究进展
谢凤,柳威,陈临溪
摘要:细胞自噬(autophagy)是将细胞内受损、变性或衰老的蛋白质以及细胞器运输到溶酶体内进行消化降解的过程。细胞自噬既是一种广泛存在的正常生理过程,又是细胞对不良环境的一种防御机制,参与多种疾病的病理过程。正常水平的自噬可以保护细胞免受环境刺激的影响,但自噬过度和自噬不足却可能导致疾病的发生。在心脏中,心肌细胞自噬对维持心肌功能具有重要的作用,自噬的异常可能导致各种心肌疾病如溶酶体储积症(Danon disease)等。各种心血管刺激如心肌缺血(ischemia)、再灌注(reperfusion)损伤、慢性缺氧(chronic hypoxia)等均可诱导心肌细胞自噬增强。而这些情况下心肌细胞自噬的作用还不清楚:它是否是一种潜在的细胞存活机制还是导致细胞死亡或疾病发生的病理性机制,或者是同时具有两种作用,目前还没有定论。心脏疾病是心肌功能出现异常时产生的各种病理状态的总称。在多种心脏疾病中,均伴随有心肌细胞自噬的改变,且影响着疾病的发生发展。在心肌肥厚
(hypertrophic cardiomyopathy)中,细胞自噬程度降低而加剧心肌肥厚;在心力衰竭(heart failure,HF)中,细胞自噬增强可导致心肌细胞自噬性死亡;而在心肌梗死(myocardial infarction,MI)中,细胞自噬增强可减小梗死面积。但是细胞自噬在心脏疾病中到底扮演着怎样的角色,取决于细胞自噬发生的水平及病理状态。目前越来越多的人开始关注药物与细胞自噬调节之间的联系,且主要集中于抗肿瘤药物及心血管调节药物的研究。另外,有报道维生素类以及雌激素受体拮抗剂他莫西芬对细胞自噬也具有调节作用。研究心肌细胞自噬与心脏疾病的关系,以及药物对细胞自噬的调节,将有利于从自噬的角度探讨心脏疾病的发生发展过程及机制,开发出治疗心脏疾病的药物。
关键词:细胞自噬;心肌功能;心血管应激;心脏疾病;药物
来源出版物:生物化学与生物物理进展, 2012, 39(3): 224-233
被引频次:23
PI3K/Akt/mTOR信号通路在巨噬细胞自噬及动脉粥样硬化斑块不稳定中的作用
王和峰,翟纯刚,庞文会,等
摘要:目的:探讨磷脂酰肌醇3-激酶(PI3K)/蛋白激酶B(Akt)/哺乳动物雷帕霉素靶蛋白(mTOR)信号通路在巨噬细胞自体吞噬以及动脉粥样硬化斑块不稳定中的作用。方法:利用Akt抑制剂康士得(20 μmol/L)、mTOR抑制剂雷帕霉素(10 nmol/L)及mTOR-siRNA(30 nmol/L)体外处理小鼠RAW 264.7巨噬细胞株48 h后,透射电镜观察巨噬细胞自噬体的变化,细胞免疫荧光法及Western blotting法检测微管相关蛋白LC3-Ⅱ表达,实时荧光定量qRT-PCR和Western blotting法检测Akt、mTOR及自噬相关蛋白Beclin 1的表达,ELISA检测巨噬细胞分泌炎症因子水平。体内实验中,24只雄性新西兰兔给予球囊损伤+1%胆固醇喂养8周,然后随机分为对照组、康士得(1.0 mg·kg-1·d-1)组和雷帕霉素(0.5 mg·kg-1·d-1)组,每组8只,干预4周。血管内超声(IVUS)检测斑块的影像学特征,透射电镜观察斑块中巨噬细胞超微结构的改变,免疫荧光法检测微管相关蛋白LC3-Ⅱ表达,免疫组织化学法检测巨噬细胞Akt和mTOR的蛋白表达。结果:与对照组比较,康士得、雷帕霉素及mTOR-siRNA干预巨噬细胞后,透射电镜下观察到自噬体明显增多,微管相关蛋白 LC3-Ⅱ和自噬相关蛋白Beclin 1的表达水平明显上调,而Akt及mTOR的mRNA及蛋白表达水平明显减少,巨噬细胞分泌的 IL-10明显降低,而 IFN-γ的分泌显著增加。体内实验:IVUS显示,与对照组比较,康士得组及雷帕霉素组的外弹性膜面积(EEMA)、斑块面积(PA)及斑块负荷(PB)明显减少,透射电镜下观察到巨噬细胞中自噬体增加,组织免疫荧光法示 LC3-Ⅱ明显增加,HE染色显示斑块纤维帽的厚度明显增加,内、中膜厚度显著减低,组织免疫组化染色显示巨噬细胞RAM-11及p-mTOR染色显著减少。结论:选择性抑制PI3K/Akt/mTOR信号通路能诱导巨噬细胞自噬,减少斑块巨噬细胞的浸润,抑制炎症反应进而稳定动脉粥样硬化易损斑块。
关键词:PI3K/Akt/mTOR信号通路;巨噬细胞;自噬;易损斑块
来源出版物:中国病理生理杂志, 2013, 29(3): 390-397
被引频次:1702
Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice
Hara, T; Nakamura, K; Matsui, M; et al.
Abstract: Autophagy is an intracellular bulk degradation process through which a portion of the cytoplasm is delivered to lysosomes to be degraded. Although the primary role of autophagy in many organisms is in adaptation to starvation, autophagy is also thought to be important for normal turnover of cytoplasmic contents, particularly in quiescent cells such as neurons. Autophagy may have a protective role against the development of a number of neurodegenerative diseases. Here we report that loss of autophagy causes neurodegeneration even in the absence of any disease-associated mutant proteins. Mice deficient for Atg5 (autophagy-related 5) specifically in neural cells develop progressive deficits in motor function that are accompanied by the accumulation of cytoplasmic inclusion bodies in neurons. In Atg5-/-cells, diffuse, abnormal intracellular proteins accumulate, and then form aggregates and inclusions. These results suggest that the continuous clearance of diffuse cytosolic proteins through basal autophagy is important for preventing the accumulation of abnormal proteins, which can disrupt neural function and ultimately lead to neurodegeneration.
来源出版物:Nature, 2006, 441(7095): 885-889
被引频次:1700
Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy
Pattingre, S; Tassa, A; Qu, XP; et al.
Abstract: Apoptosis and autophagy are both tightly regulated biological processes that play a central role in tissue homeostasis, development, and disease. The antiapoptotic protein, Bcl-2, interacts with the evolutionarily conserved autophagy protein, Beclin 1. However, little is known about the functional significance of this interaction. Here, we show that wild-type Bcl-2 antiapoptotic proteins, but not Beclin 1 binding defective mutants of Bcl-2, inhibit Beclin 1-dependent autophagy in yeast and mammalian cells and that cardiac Bcl-2 transgenic expression inhibits autophagy in mouse heart muscle. Furthermore, Beclin 1 mutants that cannot bind to Bcl-2 induce more autophagy than wild-type Beclin 1 and, unlike wild-type Beclin 1, promote cell death. Thus, Bcl-2 not only functions as an antiapoptotic protein, but also as an antiautophagy protein via its inhibitory interaction with Beclin 1. This antiautophagy function of Bcl-2 may help maintain autophagy at levels that are compatible with cell survival, rather than cell death.
来源出版物:Cell, 2005, 122(6): 927-939
被引频次:1648
Induction of autophagy and inhibition of tumorigenesis by beclin 1
Liang, XH; Jackson, S; Seaman, M; et al.
Abstract: The process of autophagy, or bulk degradation of cellular proteins through an autophagosomic-lysosomal pathway, is important in normal growth control and may be defective in tumour cells. However, little is known about the genetic mediators of autophagy in mammalian cells or their role in tumour development. The mammalian gene encoding Beclin 1, a novel Bcl-2-interacting, coiled-coil protein, has structural similarity to the yeast autophagy gene, apg6/vps30, and is mono-allelically deleted in 40%-75% of sporadic human breast cancers and ovarian cancers. Here we show using gene-transfer techniques, that beclin 1 promotes autophagy in autophagy-defective yeast with a targeted disruption of agp6/vps30, and in human MCF7 breast carcinoma cells. The autophagy-promoting activity of beclin 1 in MCF7 cells is associated with inhibition of MCF7 cellular proliferation, in vitro clonigenicity and tumorigenesis in nude mice. Furthermore, endogenous Beclin 1 protein expression is frequently low in human breast epithelial carcinoma cell lines and tissue, but is expressed ubiquitously at high levels in normal breast epithelia. Thus, beclin 1 is a mammalian autophagy gene that can inhibit tumorigenesis and is expressed at decreased levels in human breast carcinoma. These findings suggest that decreased expression of autophagy proteins may contribute to the development or progression of breast and other human malignancies.
来源出版物:Nature, 1999, 402(6762): 672-676
被引频次:1589
Loss of autophagy in the central nervous system causes neurodegeneration in mice
Komatsu, M; Waguri, S; Chiba, T; et al.
Abstract: Protein quality-control, especially the removal of proteins with aberrant structures, has an important role in maintaining the homeostasis of non-dividing neural cells. In addition to the ubiquitin-proteasome system, emerging evidence points to the importance of autophagy-the bulk protein degradation pathway involved in starvation-induced and constitutive protein turnover-in the protein qualitycontrol process. However, little is known about the precise roles of autophagy in neurons. Here we report that loss of Atg7 (autophagy-related 7), a gene essential for autophagy, leads to neurodegeneration. We found that mice lacking Atg7 specifically in the central nervous system showed behavioural defects, including abnormal limb-clasping reflexes and a reduction in coordinated movement, and died within 28 weeks of birth. Atg7 deficiency caused massive neuronal loss in the cerebral and cerebellar cortices. Notably, polyubiquitinated proteins accumulated in autophagydeficient neurons as inclusion bodies, which increased in size and number with ageing. There was, however, no obvious alteration in proteasome function. Our results indicate that autophagy is essential for the survival of neural cells, and that impairment of autophagy is implicated in the pathogenesis of neurodegenerative disorders involving ubiquitin-containing inclusion bodies.
来源出版物:Nature, 2006, 441(7095): 880-884
被引频次:1454
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy
Pankiv, S; Clausen, TH; Lamark, T ; et al.
Abstract: Protein degradation by basal constitutive
autophagy is important to avoid accumulation of polyubiquitinated protein aggregates and development of neurodegenerative diseases. The polyubiquitinbinding protein p62/SQSTM1 is degraded by autophagy. It is found in cellular inclusion bodies together with polyubiquitinated proteins and in cytosolic protein aggregates that accumulate in various chronic, toxic, and degenerative diseases. Here we show for the first time a direct interaction between p62 and the autophagic effector proteins LC3A and -B and the related γ-aminobutyrate receptor-associated protein and γ-aminobutyrate receptor-associated like proteins. The binding is mediated by a 22-residue sequence of p62 containing an evolutionarily conserved motif. To monitor the autophagic sequestration of p62- and LC3-positive bodies, we developed a novel pH-sensitive fluorescent tag consisting of a tandem fusion of the red, acid-insensitive mCherry and the acid-sensitive green fluorescent proteins. This approach revealed that p62- and LC3-positive bodies are degraded in autolysosomes. Strikingly, even rather large p62-positive inclusion bodies (2 μm diameter) become degraded by autophagy. The specific interaction between p62 and LC3, requiring the motif we have mapped, is instrumental in mediating autophagic degradation of the p62-positive bodies. We also demonstrate that the previously reported aggresome-like induced structures containing ubiquitinated proteins in cytosolic bodies are dependent on p62 for their formation. In fact, p62 bodies and these structures are indistinguishable. Taken together, our results clearly suggest that p62 is required both for the formation and the degradation of polyubiquitin-containing bodies by autophagy.
来源出版物: Journal of Biological Chemistry, 2007, 282(33): 24131-24145
被引频次:1420
The role of autophagy during the early neonatal starvation period
Kuma, A; Hatano, M; Matsui, M ; et al.
Abstract: At birth the trans-placental nutrient supply is suddenly interrupted, and neonates face severe starvation until supply can be restored through milk nutrients. Here, we show that neonates adapt to this adverse circumstance by inducing autophagy. Autophagy is the primary means for the degradation of cytoplasmic constituents within lysosomes. The level of autophagy in mice remains low during embryogenesis; however, autophagy is immediately upregulated in various tissues after birth and is maintained at high levels for 3-12 h before returning to basal levels within 1-2 days. Mice deficient for Atg5, which is essential for autophagosome formation, appear almost normal at birth but die within 1 day of delivery. The survival time of starved Atg5-deficient neonates (approximately 12 h) is much shorter than that of wild-type mice (approximately 21 h) but can be prolonged by forced milk feeding. Atg5-deficient neonates exhibit reduced amino acid concentrations in plasma and tissues, and display signs of energy depletion. These results suggest that the production of amino acids by autophagic degradation of ‘self’ proteins, which allows for the maintenance of energy homeostasis, is important for survival during neonatal starvation.
来源出版物:Nature, 2004, 432(7020): 1032-1036
被引频次:1395
Methods in mammalian autophagy research
Mizushima, N; Yoshimori, T; Levine, B
Abstract: Autophagy has been implicated in many physiological and pathological processes. Accordingly, there is a growing scientific need to accurately identify, quantify, and manipulate the process of autophagy. However, as autophagy involves dynamic and complicated processes, it is often analyzed incorrectly. In this Primer, we discuss methods to monitor autophagy and to modulate autophagic activity, with a primary focus on mammalian macroautophagy.
来源出版物:Cell, 2010, 140(3): 313-326
被引频次:1290
Parkin is recruited selectively to impaired mitochondria and promotes their autophagy
Narendra, D; Tanaka, A; Suen, DF; et al.
Abstract: Loss-of-function mutations in Park2, the gene coding for the ubiquitin ligase Parkin, are a significant cause of early onset Parkinson’s disease. Although the role of Parkin in neuron maintenance is unknown, recent work has linked Parkin to the regulation of mitochondria. Its loss is associated with swollen mitochondria and muscle degeneration in Drosophila melanogaster, as well as mitochondrial dysfunction and increased susceptibility to mitochondrial toxins in other species. Here, we show that Parkin is selectively recruited to dysfunctional mitochondria with low membrane potential in mammalian cells. After recruitment, Parkin mediates the engulfment of mitochondria by autophagosomes and the selective elimination of
impaired mitochondria. These results show that Parkin promotes autophagy of damaged mitochondria and implicate a failure to eliminate dysfunctional mitochondria in the pathogenesis of Parkinson’s disease.
来源出版物:The Journal of Cell Biology, 2008, 183(5): 795-803
被引频次:1280
p62/SQSTM1 forms protein aggregates degraded by autophagyand has a protective effect on huntingtin-induced cell death
Bjorkoy, G; Lamark, T; Brech, A ; et al.
Abstract: Autophagic degradation of ubiquitinated protein aggregates is important for cell survival, but it is not known how the autophagic machinery recognizes such aggregates. In this study, we report that polymerization of the polyubiquitin-binding protein p62/SQSTM1 yields protein bodies that either reside free in the cytosol and nucleus or occur within autophagosomes and lysosomal structures. Inhibition of autophagy led to an increase in the size and number of p62 bodies and p62 protein levels. The autophagic marker light chain 3 (LC3) colocalized with p62 bodies and coimmunoprecipitated with p62, suggesting that these two proteins participate in the same complexes. The depletion of p62 inhibited recruitment of LC3 to autophagosomes under starvation conditions. Strikingly, p62 and LC3 formed a shell surrounding aggregates of mutant huntingtin. Reduction of p62 protein levels or interference with p62 function significantly increased cell death that was induced by the expression of mutant huntingtin. We suggest that p62 may, via LC3, be involved in linking polyubiquitinated protein aggregates to the autophagy machinery.
来源出版物:The Journal of Cell Biology, 2005, 171(4): 603-614
被引频次:1236
Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease
Ravikumar, B; Vacher, C; Berger, Z; et al.
Abstract: Huntington disease is one of nine inherited neurodegenerative disorders caused by a polyglutamine tract expansion. Expanded polyglutamine proteins accumulate abnormally in intracellular aggregates. Here we show that mammalian target of rapamycin (mTOR) is sequestered in polyglutamine aggregates in cell models, transgenic mice and human brains. Sequestration of mTOR impairs its kinase activity and induces autophagy, a key clearance pathway for mutant huntingtin fragments. This protects against polyglutamine toxicity, as the specific mTOR inhibitor rapamycin attenuates huntingtin accumulation and cell death in cell models of Huntington disease, and inhibition of autophagy has the converse effects. Furthermore, rapamycin protects against neurodegeneration in a fly model of Huntington disease, and the rapamycin analog CCI-779 improved performance on four different behavioral tasks and decreased aggregate formation in a mouse model of Huntington disease. Our data provide proof-of-principle for the potential of inducing autophagy to treat Huntington disease.
来源出版物:Nature Genetics, 2004, 36(6): 585-595
·推荐论文摘要·
巨噬细胞自噬在动脉粥样硬化中的作用
许秋莲,杨阳,田野
摘要:自噬通过溶酶体依赖的降解途径维持细胞稳态。最新研究显示巨噬细胞自噬可以促进胆固醇流出,抑制炎症体活化从而抑制动脉粥样硬化进展。在进展期斑块内,巨噬细胞自噬水平降低,斑块易损性增高,极易导致斑块破裂,引起急性冠脉综合征。因此,调控巨噬细胞自噬已成为心血管领域的关注热点,深入探究其机理将为动脉粥样硬化的防治提供新思路。
关键词:自噬;巨噬细胞;胆固醇流出;炎症体;动脉粥样硬化
来源出版物:中国动脉硬化杂志, 2016, 24(1): 97-100
联系邮箱:田野,yetian@ems.hrbmu.edu.cn
支气管哮喘与细胞自噬
马龙艳,吴琦
摘要:近年来,随着人们对细胞生物学、遗传学研究的不断深入,细胞自噬在支气管哮喘(以下简称为哮喘)中的作用日趋明朗,特别在遗传因素、氧化应激和环境等方面,自噬调控可能直接影响哮喘的发生和发展,这将有望成为哮喘治疗的新方向。本文就细胞自噬、细胞自噬与哮喘的关系进行论述。
关键词:哮喘;自噬;氧化性应激;遗传学;环境
来源出版物:天津医药, 2016, 44(1): 3-4
联系邮箱:吴琦,wq572004@163.com
白藜芦醇诱导细胞自噬在神经退行性疾病进展中的作用
董雯,王蓉
摘要:细胞自噬是清除自身异常蛋白和受损细胞器的过程,在调节细胞内环境稳态、细胞生长、发育和衰老以及疾病发生发展中起重要作用。自噬功能障碍与神经退行性疾病如阿尔茨海默病、帕金森病、亨廷顿病等密切相关。这些疾病的大脑神经元内大多存在特定的病理性蛋白的异常聚集。白藜芦醇对自噬具有调节作用,能促进自噬流的发生,有效清除易形成聚集体的病理性蛋白,对神经退行性疾病具有一定的防治潜力。本文综述了白藜芦醇调节细胞自噬方面的功能及在神经退行性疾病防治的应用。
关键词:白藜芦醇;沉默信息调控因子1;去乙酰化;自噬;神经退行性疾病;聚集体
来源出版物:药学学报, 2016, 51(1): 18-22
联系邮箱:王蓉,rong_wang72@aliyun.com
不同强度不同时间耐力训练对于大鼠心肌细胞自噬发生程度的影响
马晓雯,常芸,王世强,等
摘要:目的:探讨不同强度不同时间耐力训练对大鼠心肌细胞自噬程度的影响,为运动强度和时间对心肌细胞自噬发生机制的探讨提供依据。方法:选用48只健康成年雄性SD大鼠,随机分为安静对照组、中等强度训练组和大强度训练组,每组16只。分别以15.2 m/min速度5°坡度和28 m/min速度10°坡度进行跑台训练,每周训练5天,每次训练1 h。于第8周和16周分别取各组大鼠8只进行体重称量、超声心动图检测,计算心脏重量指数(HWI);取左心室壁用于HE染色,观察心肌组织形态;制备超薄切片进行透射电镜观察,检测自噬发生程度。结果:运动训练16周后,大强度组 HWI显著低于同期对照组及同组训练8周后(P<0.05)。超声心动图显示,训练8周后,中等强度组 EF值显著高于同期对照组(P<0.05);训练16周后,中等强度组LVPWD、LVPWS、EF均显著高于同组训练8周后,大强度组 EF值显著低于同期对照组(P<0.05)。HE染色结果显示,大强度组心肌细胞损伤严重。透射电镜检测显示,长时间大强度耐力训练使自噬小体增多,自噬程度增加。结论:耐力训练可引起心肌组织细胞良好的适应性重塑,但大强度长时间耐力训练会导致心肌纤维形态异常,线粒体损伤聚变,自噬体增多等病理现象,构成心肌损伤的结构基础。
关键词:运动强度;心肌细胞;自噬;耐力训练
来源出版物:中国运动医学杂志, 2016, 35(1): 27-31
联系邮箱:常芸,changyun@ciss.cn
雷帕霉素诱导细胞自噬在衰老相关疾病中的作用
吴伯艳,刘新光,陈维春
摘要:哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是衰老和衰老相关疾病的一个关键调节因子。雷帕霉素(rapamycin,RAPA)可通过抑制
m TOR通路,诱导和促进细胞自噬的发生。细胞自噬是维持细胞内稳态的主要方式与途径,通过降解多余的、受损的及衰老的蛋白与细胞器,为细胞重建、再生和修复提供必需原料。早老症(hutchinson-gilford progeria syndrome,HGPS)患者细胞中伴随早老蛋白(progerin)的异常聚集;此外,诸如亨廷顿病、帕金森病、阿尔茨海默病等神经退行性疾病细胞内同样出现异常蛋白质的聚集,而这些异常蛋白的清除正依赖于细胞的自噬作用。由此可见,雷帕霉素是潜在的抗衰老、治疗早老症及衰老相关疾病的重要药物。该文主要阐述雷帕霉素促进细胞自噬方面的功能及在HGPS、神经退行性疾病方面的应用。
关键词:雷帕霉素;雷帕霉素靶蛋白;自噬;早老症;早老蛋白;神经退行性疾病
来源出版物:中国药理学通报, 2015, 31(1): 11-14
联系邮箱:吴伯艳,chenwchun@126.com
细胞自噬分子机制的进展
冯文之,陈扬,俞立
摘要:细胞自噬是一类依赖于溶酶体的蛋白质降解途径,在真核生物中非常保守。自噬能够感受细胞所处环境的各种信号,如氨基酸、糖等营养物质的缺乏、pH值或渗透压的改变等,使细胞做出应激反应,在恶劣环境下存活。同时,自噬过程会清除细胞内错误折叠或聚集的蛋白质,受损或老化细胞器以维持细胞内部稳态。自噬发生时,细胞内部的胞质组分被包裹在自噬体中,自噬体与溶酶体融合进行降解,产生新的小分子,如氨基酸等供细胞重新利用。一系列研究发现自噬的信号通路非常复杂,已报道有40个自噬相关蛋白(Atg蛋白)参与了自噬体的形成过程。Atg蛋白按照一定步骤发挥功能,同时相互影响,利用内膜系统构建成一个闭合的双层膜结构。将对细胞自噬研究的历史、自噬分子机制的前沿进展进行综述。
关键词:细胞自噬;自噬体;溶酶体;Atg蛋白;蛋白质降解
来源出版物:生命科学, 2015, 27(7): 859-866
联系邮箱:俞立,liyulab@mail.tsinghua.edu.cn
细胞自噬与肿瘤的关系研究进展
杨晨,李萍,梁廷明
摘要:细胞自噬(autophagy)在肿瘤的发生发展过程中扮演着非常重要的角色。自噬作用是细胞的一种自我保护机制,是真核细胞用于清除细胞内聚物及受损细胞器,进而维持细胞内稳态的一种蛋白质降解途径。从细胞自噬的类型及其形成,细胞自噬的分子调控机制,自噬对肿瘤发生及发展、以及治疗耐药等恶性行为的影响,肿瘤中自噬与预后的关联,干预自噬对肿瘤治疗的影响和细胞自噬的研究方法等方面进行综述,以期为肿瘤的治疗提供新思路。
关键词:细胞自噬;肿瘤;信号通路
来源出版物:生命科学, 2015, 27(002): 151-160
联系邮箱:梁廷明,tmliang@njnu.edu.cn
脂多糖通过PI3K/Akt/mTOR通路调控巨噬细胞自噬
杜涛,黄海,陈欣,等
摘要:目的:观察脂多糖对巨噬细胞自噬活化的影响及相关信号通路的探讨。方法:体外培养巨噬细胞株RAW264.7,分为对照组、饥饿状态激活自噬组、单纯脂多糖(LPS)刺激组、LPS+PI3K抑制剂(hVps34)组和 LPS+mTOR抑制剂(雷帕霉素)组。构建荧光真核表达载体pcDNA3.1-GFP-LC3,转染巨噬细胞,通过荧光显微镜观察各组细胞中自噬体形成情况。qRT-PCR方法检测各组中与细胞自噬相关的Atg5、Atg7、LC3-II和Bnip3 mRNA表达水平的改变。利用Western blotting检测LC3-II、p-Akt和p-mTOR蛋白在各组中的表达情况,以评价LPS激活巨噬细胞自噬的分子通路。结果:成功构建稳定表达GFP-LC3的巨噬细胞,在荧光显微镜下可以观察到自噬在饥饿状态组、LPS+hVps34组和LPS+雷帕霉素组均有明显增强;qRT-PCR检测到Atg5、LC3-II和 Bnip3 mRNA的表达在饥饿状态组、LPS+ hVps34组和 LPS+雷帕霉素组均有明显增强,而在 LPS组中略微下降;Western blotting检测发现p-Akt在饥饿状态组、LPS组和LPS+雷帕霉素组中表达明显升高;p-mTOR在饥饿状态组、LPS+hVps34组和LPS+雷帕霉素组表达明显下降;LC3-II的表达在饥饿状态组、LPS+ hVps34组和 LPS+雷帕霉素组中表达要高于对照组和LPS组。结论:LPS参与巨噬细胞自噬的调控,其可能的信号通路为PI3K/Akt/mTOR通路,但仍存在其它有效的调控通路。
关键词:脂多糖类;自噬;巨噬细胞;Akt
来源出版物:中国病理生理杂志, 2014, 30(4): 675-680
联系邮箱:陈慧,zheling76@163.com
营养缺乏对人肺癌细胞自噬的诱导作用
郭倩倩,刘志燕,姜丽丽,等
摘要:目的:观察营养缺乏对人非小细胞肺癌 A549及95D细胞自噬活性的影响,建立A549及95D细胞的自噬模型。方法:以EBSS缓冲液代替1640完全培养基,饥饿诱导处于对数生长期的A549及95D细胞0、1、2、3、4、5 h后,采用单丹磺酰戊二胺(MDC)荧光染色法检测细胞内自噬泡的形成,并采用Western blot方法检测自噬特异性基因微管相关蛋白1轻链3(LC3)和自噬相关基因Beclin1的蛋白表达水平。结果:饥饿处理A549及95D细胞后,胞内MDC荧光颗粒逐渐增多,饥饿4 h达峰值;Beclin1的表达及LC3-II与LC3-I蛋白表达量的比值(LC3-II/LC3-I)随着饥饿时间的延长逐渐增加,分别在饥饿3 h和4 h达峰值。结论:营养缺乏可以诱导增强A549及95D细胞的自噬活性,在饥饿4 h时细胞自噬水平达峰值;成功构建了营养缺乏诱导的人肺癌细胞自噬模型,为深入研究自噬在非小细胞肺癌发生发展过程中的作用及其机制奠定了良好的基础。
关键词:半导体激光器;驱动电路;恒流源
来源出版物:光电技术应用, 2013, 28(6): 71-73
细胞自噬在肝脏疾病中的作用
黄兰蔚,徐列明
摘要:近年来,越来越多的证据表明细胞自噬在慢性肝炎病毒感染、酒精性肝病、脂肪肝等各种类型的肝脏疾病的发生发展中起到重要作用,成为关注和研究的新焦点。自噬是指细胞利用溶酶体大范围降解长寿命蛋白质、大分子物质、核糖体及受损细胞器的过程。简述了各种肝脏疾病与细胞自噬的关系,认为探索细胞自噬在肝病机制中所扮演的角色,将可能成为治疗肝脏疾病的一个新靶点。
关键词:自噬;肝疾病;综述
来源出版物:临床肝胆病杂志, 2014, 2: 186-188
Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: Potential therapeutic targets for acute CNS injuries
Nakka, VP; Prakash-babu, P; Vemuganti R
Abstract: Endoplasmic reticulum (ER) stress induces a variety of neuronal cell death pathways that play a critical role in the pathophysiology of stroke. ER stress occurs when unfolded/misfolded proteins accumulate and the folding capacity of ER chaperones exceeds the capacity of ER lumen to facilitate their disposal. As a consequence, a complex set of signaling pathways will be induced that transmit from ER to cytosol and nucleus to compensate damage and to restore the normal cellular homeostasis, collectively known as unfolded protein response (UPR). However, failure of UPR due to severe or prolonged stress leads to cell death. Following acute CNS injuries, chronic disturbances in protein folding and oxidative stress prolong ER stress leading to sustained ER dysfunction and neuronal cell death. While ER stress responses have been well studied after stroke, there is an emerging need to study the association of ER stress with other cell pathways that exacerbate neuronal death after an injury. In this review, we summarize the current understanding of the role for ER stress in acute brain injuries, highlighting the diverse molecular mechanisms associated with ER stress and its relation to oxidative stress and autophagy. We also discussed the existing and developing therapeutic options aimed to reduce ER stress to protect the CNS after acute injuries.
关键词:ER stress; oxidative stress; autophagy; crosstalk; acute CNS injury
来源出版物:Molecular Neurobiology, 2016, 53(1): 532-544
联系邮箱:Vemuganti, R; vemuganti@neurosurgery.wisc.edu
CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy
Zhang, X; Han, JQ; Man, K; et al.
Abstract: Background & Aims: CXC chemokine receptor 3 (CXCR3) is involved in virus-related chronic liver inflammation. However, the role of CXCR3 in non-alcoholic steatohepatitis (NASH) remains unclear. We aimed to investigate the role of CXCR3 in NASH. Methods: Human liver tissues were obtained from 24 non-alcoholic fatty liver disease (NAFLD) patients and 20 control subjects. CXCR3 knockout (CXCR3-/-), obese db/db mice and their wild-type (WT) littermates were used in both methionine-and-choline-deficient (MCD) diet and high-fat high-carbohydrate high-cholesterol (HFHC) diet-induced NASH models. In addition, MCD-fed WT mice were administrated with CXCR3 specific antagonists. Results: CXCR3 was significantly upregulated in liver tissues of patients with NAFLD and in dietary-induced NASH animal models. Compared with WT littermates, CXCR3-/-mice were more resistant to both MCD and
HFHC diet-induced steatohepatitis. Induction of CXCR3 in dietary-induced steatohepatitis was associated with the increased expression of hepatic pro-inflammatory cytokines, activation of NF-kappa B, macrophage infiltration and T lymphocytes accumulation (Th1 and Th17 immune response). CXCR3 was also linked to steatosis through inducing hepatic lipogenic genes. Moreover, CXCR3 is associated with autophagosome-lysosome impairment and endoplasmic reticulum (ER) stress in steatohepatitis as evidenced by LC3-II and p62/SQSTM1 accumulation and the induction of GRP78, phospho-PERK and phospho-eIF2 alpha. Inhibition of CXCR3 using CXCR3 antagonist significantly suppressed MCD-induced steatosis and hepatocytes injury in AML-12 hepatocytes. Blockade of CXCR3 using CXCR3 antagonists in mice reversed the established steatohepatitis. Conclusions: CXCR3 plays a pivotal role in NASH development by inducing production of cytokines, macrophage infiltration, fatty acid synthesis and causing autophagy deficiency and ER stress.
来源出版物:Journal of Hepatology, 2016, 64(1): 160-170
联系邮箱:Yu, J; junyu@cuhk.edu.hk
Homeostatic control of innate lung inflammation by vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis
Lu, Q; Yokoyama, CC; Williams, JW; et al.
Abstract: Mutations in the autophagy gene EPG5 are linked to the multisystem human disease Vici syndrome, which is characterized in part by pulmonary abnormalities, including recurrent infections. We found that Epg5-deficient mice exhibited elevated baseline innate immune cellular and cytokine-based lung inflammation and were resistant to lethal influenza virus infection. Lung transcriptomics, bone marrow transplantation experiments, and analysis of cellular cytokine expression indicated that Epg5 plays a role in lung physiology through its function in macrophages. Deletion of other autophagy genes including Atg14, Fip200, Atg5, and Atg7 in myeloid cells also led to elevated basal lung inflammation and influenza resistance. This suggests that Epg5 and other Atg genes function in macrophages to limit innate immune inflammation in the lung. Disruption of this normal homeostatic dampening of lung inflammation results in increased resistance to influenza, suggesting that normal homeostatic mechanisms that limit basal tissue inflammation support some infectious diseases.
来源出版物:Cell Host & Microbe, 2016, 19(1): 102-113
联系邮箱:Virgin, HW; virgin@wustl.edu
The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy
Lazarou, M; Sliter, DA; Kane, LA; et al.
Abstract: Protein aggregates and damaged organelles are tagged with ubiquitin chains to trigger selective autophagy. To initiate mitophagy, the ubiquitin kinase PINK1 phosphorylates ubiquitin to activate the ubiquitin ligase parkin, which builds ubiquitin chains on mitochondrial outer membrane proteins, where they act to recruit autophagy receptors. Using genome editing to knockout five autophagy receptors in HeLa cells, here we show that two receptors previously linked to xenophagy, NDP52 and optineurin, are the primary receptors for PINK1- and parkin-mediated mitophagy. PINK1 recruits NDP52 and optineurin, but not p62, to mitochondria to activate mitophagy directly, independently of parkin. Once recruited to mitochondria, NDP52 and optineurin recruit the autophagy factors ULK1, DFCP1 and WIPI1 to focal spots proximal to mitochondria, revealing a function for these autophagy receptors upstream of LC3. This supports a new model in which PINK1-generated phospho-ubiquitin serves as the autophagy signal on mitochondria, and parkin then acts to amplify this signal. This work also suggests direct and broader roles for ubiquitin phosphorylation in other autophagy pathways.
来源出版物:Nature, 2015, 524(7565): 309-314
联系邮箱:Youle, RJ; youler@ninds.nih.gov
Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB
Medina, DL; Di, Paola S; Peluso, I; et al.
Abstract: The view of the lysosome as the terminal end of cellular catabolic pathways has been challenged by recent studies showing a central role of this organelle in the control of cell function. Here we show that a lysosomal Ca2+signalling mechanism controls the activities of the phosphatase calcineurin and of its substrate TFEB, a master transcriptional regulator of lysosomal biogenesis and autophagy. Lysosomal Ca2+release through mucolipin 1 (MCOLN1) activates calcineurin, which binds and dephosphorylates TFEB, thus promoting its nuclear translocation. Genetic and pharmacological inhibition of calcineurin suppressed TFEB activity during starvation and physical exercise, while calcineurin overexpression and constitutive activation had the opposite effect. Induction of autophagy and lysosomal biogenesis through TFEB required MCOLN1-mediated calcineurin activation. These
data link lysosomal calcium signalling to both calcineurin regulation and autophagy induction and identify the lysosome as a hub for the signalling pathways that regulate cellular homeostasis.
来源出版物:Nature Cell Biology, 2015, 17(3): 288-299
联系邮箱:Medina, DL; medina@tigem.it
Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress
Ikeda, Y; Shirakabe, A; Maejima, Y; et al.
Abstract: Rationale: Both fusion and fission contribute to mitochondrial quality control. How unopposed fusion affects survival of cardiomyocytes and left ventricular function in the heart is poorly understood. Objective: We investigated the role of dynamin-related protein 1 (Drp1), a GTPase that mediates mitochondrial fission, in mediating mitochondrial autophagy, ventricular function, and stress resistance in the heart. Methods and Results: Drp1 downregulation induced mitochondrial elongation, accumulation of damaged mitochondria, and increased apoptosis in cardiomyocytes at baseline. Drp1 downregulation also suppressed autophagosome formation and autophagic flux at baseline and in response to glucose deprivation in cardiomyocytes. The lack of lysosomal translocation of mitochondrially targeted Keima indicates that Drp1 downregulation suppressed mitochondrial autophagy. Mitochondrial elongation and accumulation of damaged mitochondria were also observed in tamoxifen- inducible cardiac-specific Drp1 knockout mice. After Drp1 downregulation, cardiac-specific Drp1 knockout mice developed left ventricular dysfunction, preceded by mitochondrial dysfunction, and died within 13 weeks. Autophagic flux is significantly suppressed in cardiacspecific Drp1 knockout mice. Although left ventricular function in cardiac-specific Drp1 heterozygous knockout mice was normal at 12 weeks of age, left ventricular function decreased more severely after 48 hours of fasting, and the infarct size/area at risk after ischemia/reperfusion was significantly greater in cardiac-specific Drp1 heterozygous knockout than in control mice. Conclusions: Disruption of Drp1 induces mitochondrial elongation, inhibits mitochondrial autophagy, and causes mitochondrial dysfunction, thereby promoting cardiac dysfunction and increased susceptibility to ischemia/reperfusion.
关 键 词 : autophagy; Drp1 protein, mouse; heart; ischemia/reperfusion injury; mitochondria
来源出版物:Circulation Research, 2015, 116(2): 264-278
联系邮箱:Sadoshima, J, sadoshju@njmsrutgers.edu
The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy
Watson, RO; Bell, SL; MacDuff, DA; et al.
Abstract: Type I interferons (IFNs) are critical mediators of antiviral defense, but their elicitation by bacterial pathogens can be detrimental to hosts. Many intracellular bacterial pathogens, including Mycobacterium tuberculosis, induce type I IFNs following phagosomal membrane perturbations. Cytosolic M. tuberculosis DNA has been implicated as a trigger for IFN production, but the mechanisms remain obscure. We report that the cytosolic DNA sensor, cyclic GMP-AMP synthase (cGAS), is required for activating IFN production via the STING/ TBK1/IRF3 pathway during M. tuberculosis and L. pneumophila infection of macrophages, whereas L. monocytogenes short-circuits this pathway by producing the STING agonist, c-di-AMP. Upon sensing cytosolic DNA, cGAS also activates cell-intrinsic antibacterial defenses, promoting autophagic targeting of M. tuberculosis. Importantly, we show that cGAS binds M. tuberculosis DNA during infection, providing direct evidence that this unique host-pathogen interaction occurs in vivo. These data uncover a mechanism by which IFN is likely elicited during active human infections.
来源出版物:Cell Host & Microbe, 2015, 17(6): 811-819
联系邮箱:Cox, JS; jeffery.cox@ucsf.edu
Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation
Wong, YC; Holzbaur, ELF
Abstract: Mitophagy is a cellular quality control pathway in which the E3 ubiquitin ligase parkin targets damaged mitochondria for degradation by autophagosomes. We examined the role of optineurin in mitophagy, as mutations in optineurin are causative for amyotrophic lateral sclerosis (ALS) and glaucoma, diseases in which mitochondrial dysfunction has been implicated. Using live cell imaging, we demonstrate the parkin-dependent recruitment of optineurin to mitochondria damaged by depolarization or reactive oxygen species. Parkin’s E3 ubiquitin ligase activity is required to ubiquitinate outer mitochondrial membrane proteins, allowing optineurin to stably associate
with ubiquitinated mitochondria via its ubiquitin binding domain; in the absence of parkin, optineurin transiently localizes to damaged mitochondrial tips. Following optineurin recruitment, the omegasome protein double FYVE-containing protein 1 (DFCP1) transiently localizes to damaged mitochondria to initialize autophagosome formation and the recruitment of microtubule-associated protein light chain 3 (LC3). Optineurin then induces auto- phagosome formation around damaged mitochondria via its LC3 interaction region (LIR) domain. Depletion of endogenous optineurin inhibits LC3 recruitment to mitochondria and inhibits mitochondrial degradation. These defects are rescued by expression of siRNA-resistant wild-type optineurin, but not by an ALS-associated mutant in the ubiquitin binding domain (E478G), or by optineurin with a mutation in the LIR domain. Optineurin and p62/SQSTM1 are independently recruited to separate domains on damaged mitochondria, and p62 is not required for the recruitment of either optineurin or LC3 to damaged mitochondria. Thus, our study establishes an important role for optineurin as an autophagy receptor in parkin-mediated mitophagy and demonstrates that defects in a single pathway can lead to neurodegenerative diseases with distinct pathologies.
关 键 词 : mitophagy; autophagosome; optineurin; amyotrophic lateral sclerosis; Parkinson’s disease
来源出版物:Proceedings of the National Academy of Sciences, 2014, 111(42): E4439-E4448
联系邮箱:Holzbaur, ELF; holzbaur@mail.med.upenn.edu
The return of the nucleus: Transcriptional and epigenetic control of autophagy
Fullgrabe, J; Klionsky, DJ; Joseph, B
Abstract: Autophagy is a conserved process by which cytoplasmic components are degraded by the lysosome. It is commonly seen as a cytoplasmic event and, until now, nuclear events were not considered of primary importance for this process. However, recent studies have unveiled a transcriptional and epigenetic network that regulates autophagy. The identification of tightly controlled transcription factors (such as TFEB and ZKSCAN3), microRNAs and histone marks (especially acetylated Lys16 of histone 4 (H4K16ac) and dimethylated H3K9 (H3K9me2)) associated with the autophagic process offers an attractive conceptual framework to understand the short-term transcriptional response and potential long-term responses to autophagy.
来源出版物:Nature Reviews Molecular Cell Biology, 2014, 15(1): 65-74
联系邮箱:Joseph, B; bertrand.joseph@ki.se
Autophagy and apoptosis: Where do they meet?
Mukhopadhyay, S; Panda, PK; Sinha, N; et al.
Abstract: Autophagy and apoptosis are two important cellular processes with complex and intersecting protein networks; as such, they have been the subjects of intense investigation. Recent advances have elucidated the key players and their molecular circuitry. For instance, the discovery of Beclin-1’s interacting partners has resulted in the identification of Bcl-2 as a central regulator of autophagy and apoptosis, which functions by interacting with both Beclin-1 and Bax/Bak respectively. When localized to the endoplasmic reticulum and mitochondria, Bcl-2 inhibits autophagy. Cellular stress causes the displacement of Bcl-2 from Beclin-1 and Bax, thereby triggering autophagy and apoptosis, respectively. The induction of autophagy or apoptosis results in disruption of complexes by BH3-only proteins and through post-translational modification. The mechanisms linking autophagy and apoptosis are not fully defined; however, recent discoveries have revealed that several apoptotic proteins (e.g., PUMA, Noxa, Nix, Bax, XIAP, and Bim) modulate autophagy. Moreover, autophagic proteins that control nucleation and elongation regulate intrinsic apoptosis through calpain- and caspase-mediated cleavage of autophagy-related proteins, which switches the cellular program from autophagy to apoptosis. Similarly, several autophagic proteins are implicated in extrinsic apoptosis. This highlights a dual cellular role for autophagy. On one hand, autophagy degrades damaged mitochondria and caspases, and on the other hand, it provides a membrane-based intracellular platform for caspase processing in the regulation of apoptosis. In this review, we highlight the crucial factors governing the crosstalk between autophagy and apoptosis and describe the mechanisms controlling cell survival and cell death.
关键词:autophagy; apoptosis; crosstalk; Bcl-2; Beclin-1; BH3-only proteins
来源出版物:Apoptosis, 2014, 19(4): 555-566
联系邮箱:Bhutia, SK; sujitb@nitrkl.ac.in
编辑:王微
Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae
Tsukada, M; Ohsumi, Y
Autophagy in the yeast is similar to that in mammalian cells. A mutant designated as apg1 (autophagy) defective in accumulation of autophagic bodies in the vacuoles was isolated by selection using a light microscope from a mutagenized proteinase-deficient strain. In the apg1 strain, which has normal vacuolar proteinases, nitrogen starvation did not induce protein degradation. The apg1 mutant lost its viability faster than wild-type cells during nitrogen starvation. By using the loss of viability as a first screening test, 75 other apg mutants were selected. These apg mutants including apg1 fell into 15 complementation groups. Genetic analyses of representative apg mutants revealed that they all had single recessive chromosomal mutations. Strains with each apg mutation were defective in protein degradation in the vacuoles induced by nitrogen starvation and homozygous diploids for each apg mutation did not sporulate. These results on the apg mutants suggest that autophagy via autophagic bodies is indispensable for protein degradation in the vacuoles under starvation conditions, and that at least 15 APG genes are involved in autophagy in yeast.
猜你喜欢
现代临床医学(2022年3期)2022-06-06 07:59:58
好日子(2021年8期)2021-11-04 09:02:52
中老年保健(2021年2期)2021-08-22 07:29:12
家庭医学(下半月)(2020年1期)2020-05-11 02:05:32
海峡姐妹(2018年7期)2018-07-27 02:30:36
特别健康(2018年4期)2018-07-03 00:38:08
特别健康(2018年2期)2018-06-29 06:13:42
新闻传播(2016年17期)2016-07-19 10:12:05
健康管理(2015年4期)2015-11-20 23:22:29
发明与创新(2015年37期)2015-02-27 10:40:34
