
基于高通量测序的野生毛葡萄转录组数据分析
2016-12-08 19:03方辉蒋胜理曲俊杰周思泓
湖北农业科学 2016年18期
方辉++蒋胜理++曲俊杰++周思泓


摘要:利用RNA-seq技术对所构建的野生毛葡萄(Vitis quinquangularis Rehd)叶片的转录组进行测定,对原始reads进行过滤和组装,得到了35 238条质量较高的unigene,平均长度为1 081 nt,N50为1 735 nt。基于NCBI蛋白质数据库(Nr)、蛋白质序列数据库(Swiss-Prot)、京都基因与基因组百科全书(KEGG)数据库和直系同源基因簇(COG)进行相似性比对,共注释了26 751条unigene,另有8 487条unigene未被注释。物种同源性显示与葡萄的同源性最高为74.48%。利用COG数据库将unigene分成25类,通过GO分类和KEGG富集性分析,将unigene分别归类于44个GO类别和122个代谢途径。此外,在35 238条unigene中共搜索到4 428个SSR位点,二核苷酸的SSR数目最多(1 906条),其次为三核苷酸(1 762条)。这些信息为毛葡萄功能基因、相关候选基因的发掘以及分子标记辅助育种提供了重要依据。
关键词:毛葡萄(Vitis quinquangularis Rehd);转录组;高通量测序
中图分类号:S663.1 文献标识码:A 文章编号:0439-8114(2016)18-4827-05
DOI:10.14088/j.cnki.issn0439-8114.2016.18.050
野生毛葡萄(Vitis quinquangularis Rehd)是葡萄科(Vitaceae)葡萄属(Vitis)真葡萄亚属(Euvitis)东亚种群,盛产于中国,是中国葡萄属东亚种群中分布最广的一个野生种[1]。广西壮族自治区作为中国野生毛葡萄之乡和中国野生毛葡萄酒原产地,在桂中、桂北、桂西的喀斯特地貌地区野生毛葡萄较集中。野生毛葡萄含多种维生素、各种氨基酸、矿物质等营养物质,还含有丰富的花色素双糖苷、超氧化歧化酶(SOD)及白藜芦醇(RES)物质,具有软化血管、抗衰老、防癌的作用[2]。多数野生毛葡萄表现抗逆性强、耐病性好、耐南方高温多雨潮湿天气、耐旱耐瘠等优良特性,是南方石漠化治理、生态重建的优良树种,具有重要的经济价值[3,4]。目前对毛葡萄的研究主要集中在栽培技术和杂交育种方面,缺乏对其分子生物学方面的研究,对其基因信息的研究极少。
转录组是在特定发育阶段和一定生理条件下,细胞内所有转录出来的RNA的集合。通过高通量的转录组分析,可以获得机体在生命过程中基因的表达模式[5]。RNA-seq是2008年建立起来的基于深度测序的转录组分析技术,它能够在单核苷酸水平上对任何物种进行整体转录活动的检测,是当前在全基因组水平上研究基因表达模式的主要技术[6]。本研究拟采用RNA-seq技术对所构建的毛葡萄叶片的转录组进行测定,并在基因组水平上进行转录组分析,旨在获得毛葡萄更多的转录本和更为全面的转录组信息,为重要性状相关基因的克隆及功能分析、鉴定次级代谢物生物合成相关基因奠定基础。
1 材料与方法
1.1 材料
植物材料为毛葡萄五峒-1,生长于广西壮族自治区河池市都安瑶族自治县高岭镇五峒村,树龄数十年、生长旺盛,叶片生长状况良好,果实产量高,经调查与试验发现其对多种病害高抗或免疫。采集后液氮速冻,并转移至-80 ℃水箱保存备用。
1.2 方法
1.2.1 野生毛葡萄测序 采取野生毛葡萄的嫩叶并提取总RNA,送至北京诺禾致源生物信息科技有限公司进行文库构建与高通量测序,测序平台为Illumina HiSeq TM 2000,每个样品测序量约为4.75 G。
1.2.2 测序数据的组装 对测序后得到的原始数据total raw reads进行质量分析,去除重复、含接头、测序质量低的reads,获得clean reads。使用短reads组装软件Trinity做转录组从头组装[7]。首先将具有一定长度重叠的reads连成更长的片段,通过reads重叠关系得到组装片段contig。然后将reads比对回contig,通过paired-end reads确定来自同一转录本的不同contig以及这些contig间的距离,Trinity软件将这些contig连在一起,最后得到两端不能再延长的序列,即为unigene。
1.2.3 功能注释 利用Blastx将unigene序列与Nr(Non-redundant Protein Sequence Database in GenBank)、Swiss-Prot(Swiss-Prot Protein Sequence Database)、KEGG(Kyoto Encyclopedia of Genes and Genomes)和COG(Cluster of Orthologous Groups of proteins)数据库进行比对,获取与unigene具有最高序列相似性的蛋白,从而得到该unigene的蛋白功能注释信息。根据Nr注释信息,使用Blast2GO[8]软件进行GO注释,得到每个unigene的GO信息后,用WEGO[9]软件进行GO功能分类统计。
按照Nr、Swiss-Prot、KEGG、COG的优先级顺序,将unigene序列与以上蛋白库进行Blastx比对,取比对结果中等级最高的蛋白确定该unigene的编码区序列,然后根据标准密码子表将编码区序列翻译成氨基酸序列,从而得到该unigene编码区的核酸序列(序列方向5′→3′)和氨基酸序列。最后,将与以上蛋白库都比对不上的unigene再用软件ESTScan预测其编码区,得到其编码区的核酸序列(序列方向5′→3′)和氨基酸序列[10]。
1.2.4 SSR位点的筛选 利用MIcroSAtellite(MISA) 软件在所有unigene中搜索SSR位点,设置二核苷酸重复次数至少为6次,三核苷酸、四核苷酸、五核苷酸和六核苷酸重复次数至少为4次。
2 结果与分析
2.1 转录组测序和组装
通过Illumina Hiseq2000平台测序,总计产出56 374 280条reads,去除低质量和含有接头的reads后,得到53 697 490条clean reads,共计5 369 749 000个核苷酸(Nucleotides,nt),GC含量45.52%、Q20为95.63%。利用Trinity软件对这些reads进行组装得到35 238条unigene,平均长度为1 081 nt,N50为 1 735 nt。unigene的长度分布(图1)显示,长度大于1 000 nt的unigene 13 651条,占全部unigene的38.74%。这些说明本研究中转录组文库的测序和组装结果都较好,能够进行后续生物信息学分析。
2.2 功能注释
为了预测unigene功能,分别将unigene与Nr、Swiss-Prot、COG、KEGG等生物学数据库进行比对。通过Blast比对(表1),共有26 751条unigene获得了基因注释,占所有unigene的75.92%;有8 487条unigene(24.08%)未被注释,这些未被注释的基因可能认为是新基因。
利用blastx将组装出来的unigene序列与Nr数据库进行比对后,取每个unigene在Nr库中比对结果最好(E值最低)的那一条序列为对应同源序列(如有并列,取第一条)确定同源序列所属物种,统计比对到各个物种的同源序列数量,共找到26 719个unigene与其他近缘生物的已知基因具有不同程度的同源性,其中有19 901条(74.48%)与葡萄(Vitis vinifere)同源;1 371条(5.13%)与可可(Theobroma cacao)序列同源;865条(3.24%)与树棉(Gossypium arboreum)序列同源。35 238条unigene中还有8 519条(24.18%)unigene没有比对到近源物种 (图2)。
将所有unigene基因与COG和GO数据库进行比对,并进行功能注释与分类。26 816条unigene被注释到25个COG类别中(图3),其中一般功能基因(Generalfunction prediction only)是最大类别,包含5 280条unigene,占被注释unigene总数的19.69%; 其次是信号传导机制(Signal transduction mechanisms),包含3 293条(12.28%)unigene,其余依次为翻译后修饰,蛋白质周转以及分子伴侣类(Posttranslational modification,protein turnover,chaperones)(2 980条,11.11%),RNA加工与修改(RNA processing and modification)(1 591条,5.93%),而细胞运动(Cell motility)是最小的类别,仅包含10条unigene。
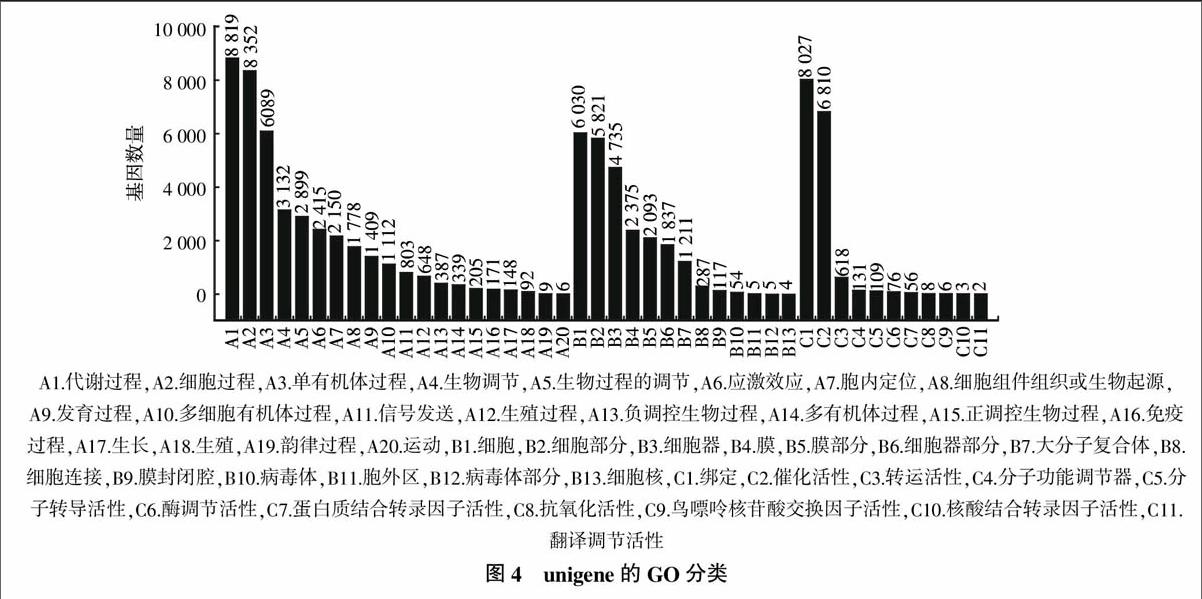
在已经得到的Nr注释信息基础上,采用Blast2GO获得毛葡萄unigene的GO分类信息,共有14 692条unigene得到GO注释(图4)。在GO分类体系中,生物学过程(biological process)、细胞组分(cellular component)和分子功能(Molecular function)。这3个大的类别又被划分为详细的44个小类别,其中“代谢过程(Metabolic process)”(8 819, 60.03%)、“细胞过程(Cellular process)”(8 352, 56.84%)和“结合(Binding)”(8 027,54.64%),3个类群占了主要部分,而“翻译调节活性(Translation regulator activity)”(2,0.01%)、“核酸结合转录因素活性(Nucleic acid binding transcription factor activity)”(3,0.02%)和“内核(nucleoid)”(4,0.04%)仅有非常少的基因归入,这一分类结果显示了毛葡萄基因表达谱的总体情况。
2.3 代谢通路分析
对野生毛葡萄叶片的转录组进行KEGG分析发现,有6 871条unigene注释到KEGG数据库中,分布于122条已知的通路中,包括淀粉和蔗糖代谢(Starch and sucrose metabolism)(165条,ko00500)、植物病原物相互作用(Plant-pathogen interaction)(145条,ko04626)和光合生物碳固定(Carbon fixation in photosynthetic organisms)(78条,ko00710)。注释序列数目较多的5个通路分别是代谢途径(Metabolic pathways)(1 743条,ko01100)、次生代谢物生物合成(Biosynthesis of secondary metabolites)(812条,ko01110)、核糖体(Ribosome)(278条,ko03010)、植物激素信号转导(Plant hormone signal transduction)(220条,ko04075)和真核细胞核糖体合成(Ribosome biogenesis in eukaryotes)(209,ko03008)(表2)。
2.4 SSR分析
利用MISA软件在毛葡萄的35 238条unigene中共搜索到4 428个SSR位点,占unigene总序列数的12.57%,平均每8.6 kb出现1个SSR,其中包含有两个及两个以上SSR的unigene共有576条。二核苷酸和三核苷酸重复类型的SSR数目最多,其中二核苷酸1 906条、三核苷酸1 762条;而五核苷酸和六核苷酸重复类型在毛葡萄中转录组序列中含量较少,仅有165条和201条(表3)。除此之外,不同核苷酸的重复次数也有很大的变化。在检测到的SSR中出现频率最高的5类基序为AG/CT 1 425个、AAG/CTT 552个、AT/TA 345个、ATC/ATG 247个、AGC/CTG 235个,上述SSR特征分析有助于开展毛葡萄及其同属物种的基因组差异分析通用性标记开发和遗传图谱构建的研究。
3 讨论
基于高通量测序技术的转录组学研究是一种高效、可靠的发掘功能基因的手段,Illumina高通量测序的数据量大、速度快、成本低、效率高[11]。本研究应用Illumina高通量测序技术对毛葡萄转录组进行测序,对毛葡萄转录组测序获得了56 374 280个reads序列,去除低质量的和含有接头的reads以后,得到53 697 490条clean reads,5 369 749 000个核苷酸,GC含量45.52%、Q20为95.63%。利用Trinity软件对这些reads进行组装得到35 238条unigene,长度从224 nt到15 540 nt,平均长度为1 081 nt,N50 为1 735 nt,N50值越大反映组装得到的长片段越多组装效果就越好。测序数据产量和数据组装质量是评价转录组测序完成情况的重要指标。以上研究结果表明,此次序列组装的质量和长度可以满足转录组分析的基本要求。
结合生物信息学分析方法对毛葡萄unigene与Nr、Swiss-Prot、COG、KEGG数据库进行比对,进行序列相似性和功能注释分析。26 719个unigene与其他近缘生物的已知基因具有不同程度的同源性,有19 901条(74.48%)与葡萄同源,与葡萄的亲缘关系最近。KEGG分析中,19 274条unigene被注释到128个代谢通路。根据KEGG数据库对上述unigene进行代谢途径分析,涉及112个具体的代谢途径分支,参与到毛葡萄体内的碳水化合物代谢、脂类代谢、次生物质代谢等过程中,为进一步大量挖掘毛葡萄生长发育过程中的重要表达基因,开展毛葡萄的基因克隆及功能验证等研究提供了基础数据。转录组数据分析结果表明,毛葡萄叶片为植物生长提供了物质、能量、信号转导以及抗病条件。
SSR分子标记具有操作简便、重复性好、多态性丰富、遗传信息量大、共显性遗传等优点,已在遗传多样性分析、遗传图谱构建、功能基因发掘、分子标记辅助育种等研究中得到了广泛应用。采取实验室方法开发SSR标记费时、耗力、成本高、试验复杂,基于转录组数据信息进行SSR分子标记开发是一种经济有效的方法。目前毛葡萄可利用的分子标记数量非常有限,转录组产生的海量数据为SSR分子标记的开发提供了丰富和极有价值的可利用资源。本研究在35 238条unigene中共搜索到4 428个SSR位点,占unigene总序列数的12.57%。二核苷酸和三核苷酸重复类型的SSR数目最多,其中二核苷酸1 906条(43.04%)、三核苷酸1 762条(39.79%),前人的研究结果显示有些物种二核苷酸占优势,另一些物种三核苷酸占优势,这种现象的出现与MISA搜索时相关参数的设置有很大的关系[12],后续可对这些SSR进行引物设计和扩增检测,筛选出扩增稳定、条带清晰、多态性好的引物为进一步开发新的SSR标记奠定了基础。
本研究采用Illumina Hiseq2000高通量测序技术建立了毛葡萄转录组数据库,获得了大量的转录本信息,并对表达基因进行了序列组装、功能注释、代谢途径等分析,为今后更深入研究毛葡萄功能基因组、基因克隆及抗逆机理研究提供了极大的方便。
参考文献:
[1] 林 玲,张 瑛,卢 江,等.中国葡萄属野生资源毛葡萄的研究与利用进展[J].中外葡萄与葡萄酒,2015(2):66-69.
[2] 涂 佳,邓学良,周文化,等.野生毛葡萄常规营养成分和原花青素含量分析[J].食品与机械,2010(2):82-85.
[3] 邹 瑜,吴代东,张 瑛,等.毛葡萄霜霉病防治试验[J].广西农业科学,2002(6):315-317.
[4] 邹 瑜. 野生毛葡萄种质资源及利用研究进展[J].南方农业学报,2008,39(5):664-667.
[5] WANG Z,GERSTEIN M,SNYDER M. RNA-Seq:A revolutionary tool for transcriptomics[J].Nature Reviews Genetics,2009, 10(1):57-63.
[6] COSTA V,ANGELINI C,DE FEIS I,et al. Uncovering the complexity of transcriptomes with RNA-Seq[J]. Journal of Biomedicine and Biotechnology,2010:853916.
[7] HAAS B J,PAPANICOLAOU A,YASSOUR M,et al. De novo transcript sequencere construction from RNA-seq using the Trinity platform for reference generation and analysis[J].Nature Protocols,2013,8(8):1494-1512.
[8] CONESA A,G?魻TZ S,GARC?魱A-G?譫MEZ J M,et al. Blast2GO:A universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21(18):3674-3676.
[9] YE J,FANG L,ZHENG H,et al. WEGO:A web tool for plotting GO annotations[J].Nucleic Acids Research,2006,34(1):293-297.
[10] BIAO J,DASEN X,WENRUI L,et al. De Novo assembly and characterization of the transcriptome, and development of SSR markers in wax gourd(Benicasa hispida)[J].Plos One,2013, 8(8):653-661.
[11] SUN C, LI Y,WU Q,et al. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis[J]. BMC Genomics,2010,11:262.
[12] WEI W L,QI X Q,WANG L H,et al. Characterization of the sesame(Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers[J].BMC Genomics,2011,12:451.
猜你喜欢
国际太空(2023年1期)2023-02-27
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
阅读(低年级)(2020年11期)2020-12-28
女报(2020年10期)2020-11-23
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
中国发展观察(2017年8期)2017-04-26
癌变·畸变·突变(2014年2期)2014-03-01
