
S4N4和Se4N4中的跨空同共轭与同芳香性
2016-12-07 05:46:16陈秀梅
厦门大学学报(自然科学版) 2016年6期
曾 慧,陈秀梅,吕 鑫
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,福建省理论与计算化学重点实验室,福建 厦门361005)
S4N4和Se4N4中的跨空同共轭与同芳香性
曾 慧,陈秀梅,吕 鑫*
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,福建省理论与计算化学重点实验室,福建 厦门361005)
通过量子化学计算从理论上证明了具有奇特双楔形笼状结构的S4N4和Se4N4分子具有同芳香性,其芳香性源于分立的NXN(X=S,Se)结构组元间极强的跨空同共轭效应.
双叶同芳香性;跨空同共轭效应;电子离域;虚核化学位移
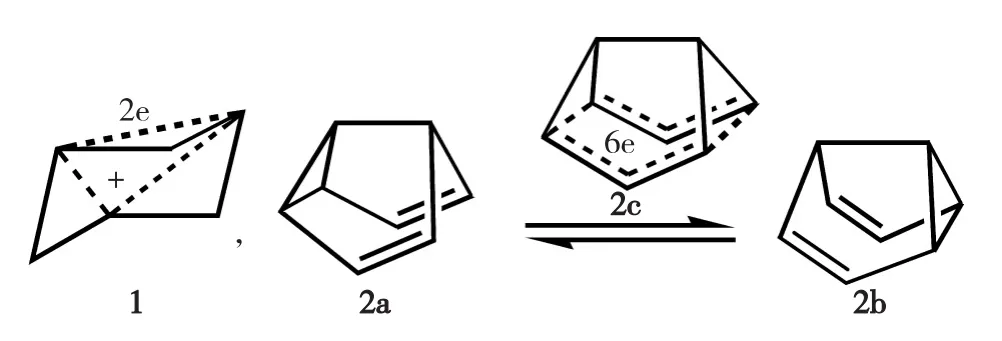
图1 具2e同共轭的3,5-双脱氢环己烷基正离子1和具6e同共轭的半瞬烯2柯普重排过渡态2cFig.1 2e-homoconjugation in tris-homocyclopropenyl cation 1 and 6e-homoconjugation in the transition state 2c for cope rearrangement of semibullvalene 2

图2 1,5-二膦-二硫化四氮分子及其2个V形NSN碎片上pπ轨道所形成的6c-10e跨空同共轭效应Fig.2 1,5-diphosphadithiatetrazocines and the 6c-10e through-space homoconjugation formed by the pπorbitals of its V-shaped NSN subunits
1959年,Winstein首次提出同芳香性(homoaromaticity)概念[1],用于描述某些化合物其共轭性被破坏后但仍呈现芳香性的特征.以3,5-双脱氢环己烷基正离子1[2]为例(图1),被3个饱和亚甲基间隔开的3个sp2杂化碳原子的pπ轨道仍能构成三中心-双电子(3c-2e)的跨空同共轭(through-space homoconjugation),满足(4N+2)休克尔规则,这种由非连续环状同共轭效应产生的芳香性即称同芳香性,又因该同共轭由3个分立不饱和基团构成而称为三叶同芳香性(trishomoaromaticity).此后,人们发现为数甚多的有机物(阳、阴)离子具有同芳香性[3-4];一些有机周环反应(如狄尔斯-阿尔德反应、1,3-偶极环加成反应、柯普重排等)的活化能较低,也可由其过渡态具有同芳香性来解释[5-6],例如,半瞬烯2柯普重排过渡态2c因其六中心-六电子(6c-6e)跨空同共轭而具有双叶同芳香性(图1).
值得注意的是,中性稳定化合物具有同芳香性的实例极为罕见[3-4,7],理论设计中性同芳香性化合物则偶见报道[8-13].本课题组之前将同芳香性概念拓展到一系列无机簇合物离子(如[SeI2+]2、S82+和[I2+]2等)和一些P、N、S杂环分子[14-16],其中1,5-二膦-二硫化四氮(1,5-diphosphadithiatetrazocine)环状分子中2个V形NSN亚单元均具三中心-五电子(3c-5e)π共轭,二者跨空形成六中心-十电子(6c-10e)同共轭体系,满足(4N+2)休克尔规则,因而具双叶同芳香性(图2)[15-16].注意到具有双重折叠环结构(亦称为双楔形笼状结构)的X4N4(X=S,Se)分子3和4中均具有类似的V形NXN(X=S,Se)亚单元及其间距[17-18](图3),由此推测这类分子中也可能存在跨空同共轭和同芳香性,本文中将通过量子化学计算加以证明.
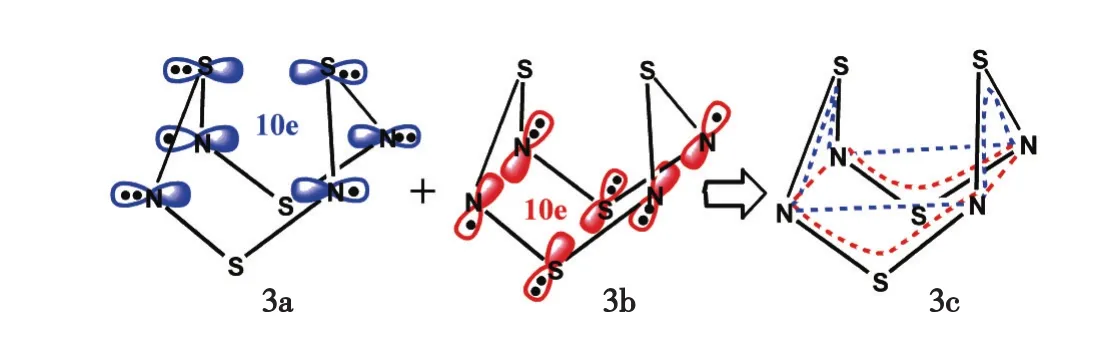
图3 S4N4中的2套6c-10e跨空同共轭效应Fig.3 Two equivalent sets of 6c-10e throughspace homoconjugation within S4N4
1 结果与讨论
1.1双楔形笼状分子X4N4(X=S,Se)的几何结构与芳香性
X射线晶体结构衍射及电子衍射实验[17-18]明确了X4N4(X=S,Se)分子具有双楔形笼状结构(图3),分子对称性属D2d点群,可看成是八元环在S原子处对折后形成双重折叠结构.其奇特性在于:1)N原子均为二配位,S—N键长均为0.162 nm(Se—N键长0.179 nm),且比常规S—N单键(约0.177 nm)短得多(常规Se—N单键长约0.185 nm),说明X—N间存在一定强度的π键;2)跨环S…S间距为0.259 nm (Se…Se间距为0.275 nm),明显短于相应原子间范德华半径之和,又明显长于S—S(或Se—Se)单键,说明跨环X…X间并非简单X—Xσ键.因此吸引了大量理论研究[19-23],但未见关于此类结构芳香性的讨论.
借鉴1,5-二膦-二硫化四氮体系中的跨空同共轭模型(图2),仅考虑S4N4中π类型价层原子轨道和电子,如图3所示,该分子上半部2个分立的NSN亚单元各含1套3c-5e的π共轭,通过跨空轨道相互作用,组成了1套6c-10e的跨空同共轭体系3a,满足(4N+2)休克尔规则,因而可能具有双叶同芳香性;类似地,该分子下半部也可形成1套6c-10e的跨空同共轭体系3b.因此,S4N4(或Se4N4)可能有两重双叶同芳香性.
B3LYP/6-311+G(3df)[24-26]计算优化的X4N4(X =S,Se)分子结构如图4所示,跨环X…X间距的理论计算预测值(S…S 0.275 nm,Se…Se 0.289 nm)略长于实验观测值,其他结构参数的理论预测值与实验观测值一致,说明该方法能合理预测分子内跨环相互作用.结合GIAO(gauge-independent atomic orbital)方法[27]计算得到分子体心位置(即N4正方形中心)处虚核化学位移(nucleus-independent chemical shift, NICS)[28-29]值分别为-31.3(S4N4)和-31.5 (Se4N4),证明该类分子确因跨空同共轭效应而具有极强芳香性.更为有趣的是,NICS极值点并不在体心位置,而是在距离N4正方形平面中心之上、下0.05 nm处(图4),这是因为2套交错的6c-10e跨空同共轭体系正好是以N4平面为分界线.ACID(anisotropy of the current-induced density)分析[30]也表明,该类分子在外磁场作用下可产生极强的抗磁环流(图5).
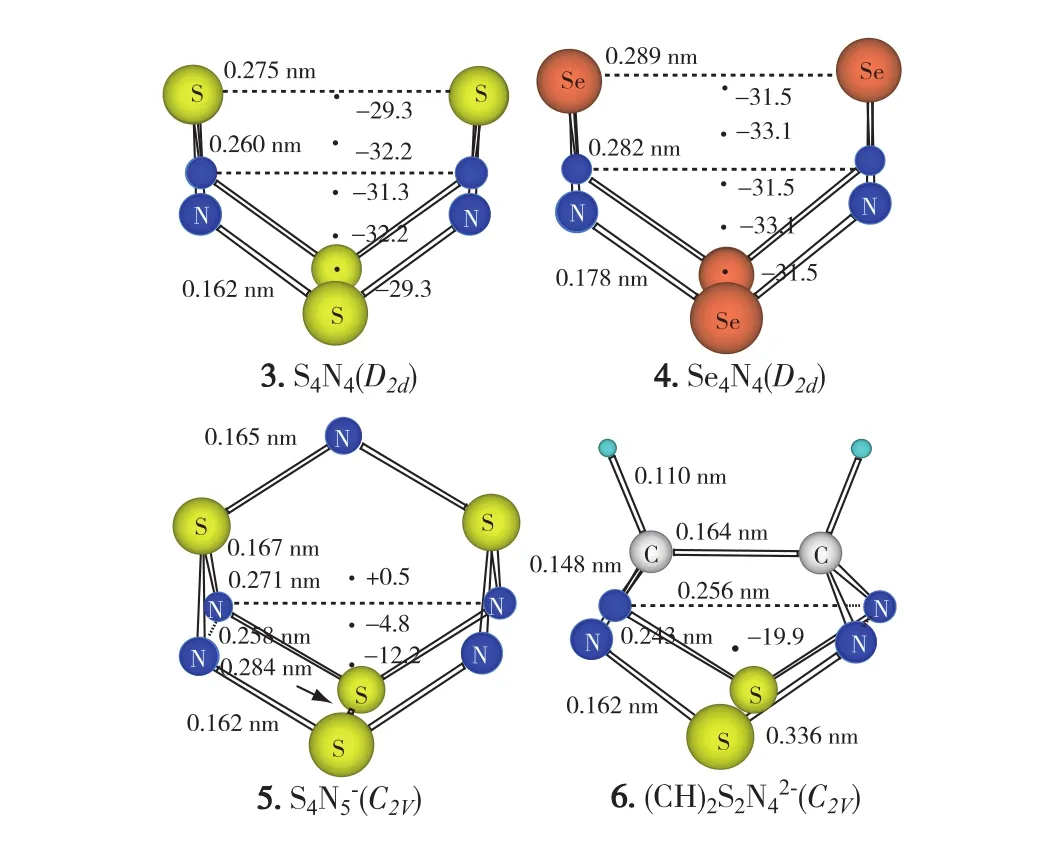
图4 B3LYP/6-311+G(3df)计算预测的S4N4、Se4N4、S4N5-和(CH)2S2N42-结构和GIAO-NICS值Fig.4 Geometries and GIAO-NICS values of S4N4, Se4N4,S4N5-and(CH)2S2N42-predicted at the B3LYP/6-311+G(3df)level of theory
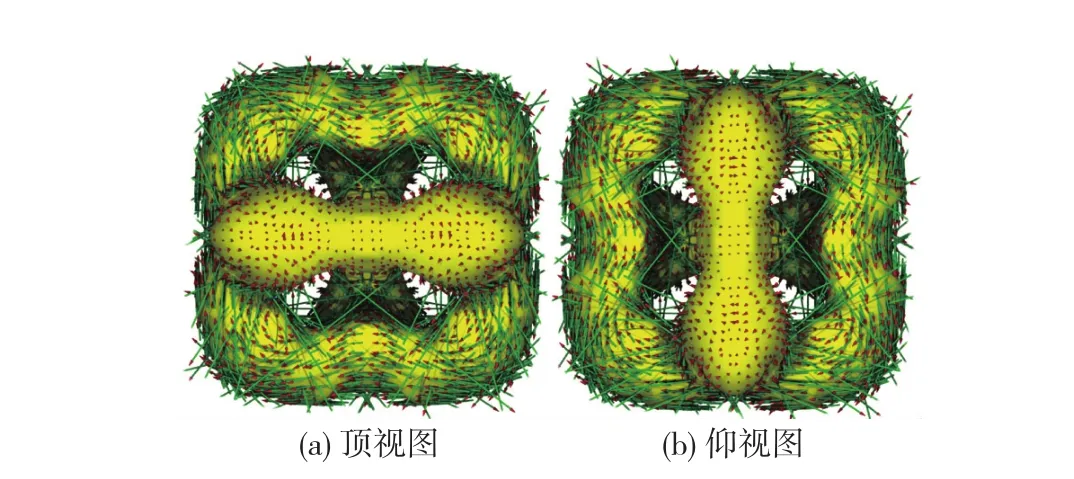
图5 S4N4(D2d)中抗磁环流的ACID分析Fig.5 Diatropic ring currents within S4N4(D2d)revealed by ACID analysis
本文中还考察了2个模型化合物S4N5-(5)[31]和(CH)2S2N42-(6),它们与1,5-二膦-二硫化四氮分子相似,均包含1套具6c-10e跨空同共轭的(NSN)2结构组件,NICS(0)值分别为-12.2和-19.9,具同芳香性.X4N4分子的电子离域效应和芳香性更强,导致其N原子相当高的负电荷(0.58左右)以及更短的跨环X…X(X=S,Se)间距(图4).
1.2X4N4(X=S,Se)的其他构型与芳香性
X4N4分子还可能取船式构型[19,32].计算表明,船式构型也存在一定程度的跨环X…X(X=S,Se)相互作用(图6);S4N4的船式构型7与双楔形笼式构型能量接近,Se4N4的船式构型8则比双楔形笼式构型能量高53.1 kJ/mol.实际上,已经发现在金属配合物中存在S4N4船式构型[32],但尚无船式构型Se4N4的实验证据.

图6 B3LYP/6-311+G(3df)计算预测的X4N4(X=S,Se)船式构型和NICS(0)值Fig.6 Geometries and NICS(0)values of the boat conformations of X4N4(X=S,Se) predicted at the B3LYP/6-311+G(3df)level of theory
两化合物船式构型中,对位的X…X间距和N…N间距均明显长于对应的原子范德华半径和,因此不会有如3b所示的(NXN)2跨空同共轭效应;但是,船式结构中除2个X—N键长接近X—N单键外,其余的X—N键长均明显短于X—N单键,说明仍存在共轭效应,其最短的间位X…X间距(最短的间位S…S间距约0.269 nm、Se…Se间距约0.293 nm,图6)也显示间位X…X跨空作用的存在.两化合物船式构型的NICS(0)值分别为-6.3和-8.2,呈弱芳香性,可能源于其2个具3c-5eπ共轭的XNX结构组件间较弱的跨空共轭效应.
X4N4的平面构型因有12个π电子而具反芳香性,其单重态NICS(0)值为+42.6.
2 结 论
环状分子X4N4(X=S,Se)双重折叠结构的电子离域本质存在争议,更未见关于其是否具有芳香性的文献报道.本研究从理论上证明了此类分子中分立的NXN结构组元具有3c-5eπ共轭,可以通过两两间的跨空轨道相互作用,形成2套交错的6c-10e跨空同共轭体系,均满足(4N+2)休克尔规则,因而具有极强同芳香性.
[1] WINSTEIN S.Homo-aromatic structures[J].J Am Chem Soc,1959,81(24):6524-6525.
[2] WINSTEIN S,SONNENBERG J,DEVRIES L.The trishomocyclopropenyl cation[J].J Am Chem Soc,1959,81 (24):6523-6524.
[3] WILLIAMS R V.Homoaromaticity[J].Chem Rev,2001, 101(5):1185-1204.
[4] WILLIAMS R V.Semibullvalenes and related molecules: ever closer approaches to neutral homoaromaticity[J]. Eur J Org Chem,2001,2001(2):227-235.
[5] ZIMMERMAN H E,GRUNEWALD G L.The chemistry of barrelene.III.A unique photoisomerization to semibullvalene[J].J Am Chem Soc,1966,88(1):183-184.
[6] ZIMMERMAN H E,BINKLEY R W,GIVENS R S,et al.Mechanistic and exploratory organic photochemistry. XLIV.The barrelene to semibullvalene transformation. Correlation of excited-state potential energy surfaces with reactivity[J].J Am Chem Soc,1969,91(12):3316-3323.
[7] WILLIAMS R V,KURTZ H A.Homoaromaticity[J]. Adv Phys Org Chem,1994,29:273-331.
[8] TANTILLO D J,HOFFMANN R,HOUK K N,et al. Extended barbaralanes:sigmatropic shiftamers orσ-polyacenes?[J].J Am Chem Soc,2004,126(13):4256-4263.
[9] WU H S,JIAO H,WANG Z X,et al.Neutral bishomoaromatic semibullvalenes[J].J Am Chem Soc,2003,125 (35):10524-10525.
[10] BROWN E C,HENZE D K,BORDEN W T.Are 1,5-disubstituted semibullvalenes that have C2vequilibrium geometries necessarily bishomoaromatic?[J].J Am Chem Soc,2002,124(50):14977-14982.
[11] GOREN A C,HROVAT D A,SEEFELDER M,et al. The search for bishomoaromatic semibullvalenes and barbaralanes:computational evidence of their identification by UV/Vis and IR spectroscopy and prediction of the existence of a blue bishomoaromatic semibullvalene[J].J Am Chem Soc,2002,124(13): 3469-3472.
[12] JIAO H,NAGELKERKE R,KURTZ H A,et al.Annelated semibullvalenes:a theoretical study of how they "cope"with strain[J].J Am Chem Soc,1997,119(25): 5921-5929.
[13] HOFFMANN R,STOHRER W D.The cope rearrangement revisited[J].J Am Chem Soc,1971,93(25): 6941-6948.
[14] ZHANG Q,LU X,HUANG R B,et al.Pericyclic transi-tion-state-like aromaticity in the inorganic ions Se2I42+and S2O42-[J].Inorg Chem,2006,45(6):2457-2460.
[15] ZHANG Q,YUE S P,LU X,et al.Homoconjugation/ homoaromaticity in main group inorganic molecules[J]. J Am Chem Soc,2009,131(28):9789-9799.
[16] CHIVERS T,HILTS R W,JIN P,et al.Synthesis,properties and bishomoaromaticity of the first tetrahalogenated derivative of a 1,5-diphosphadithiatetrazocine:a combined experimental and computational investigation [J].Inorg Chem,2010,49(8):3810-3815.
[17] DELUCIA M L,COPPENS P.Crystal structure of tetrasulfur tetranitride(S4N4)at 120 K[J].Inorg Chem, 1978,17(8):2336-2338.
[18] BÄRIGHUASEN H,VON VOLKMANN T,JANDER J.Die kristallstruktur von tetrastickstofftetraselenidN4Se4[J].Acta Crystallogr,1966,21:571-577.
[19] CHUNG G,LEE D.Tetrasulfur tetranitride and its selenium analogs:ab initio and DFT calculations[J].J Mol Struct:THEOCHEM,2002,582(1/2/3):85-90.
[20] SUONTAMO R J,LAITINEN R S.Ab-initio molecularorbital study of SenS4-nN4(n=0-4)[J].J Mol Struct: THEOCHEM,1995,336(1):55-60.
[21] MILLEFIORI S,MILLEFIORI A.Electronic states of nitrogen-sulfur compoundsN2S2andN4S4[J].Inorg Chim Acta,1980,45(1):L19-L22.
[22] MILLEFIORI A,MILLEFIORI S.Abinitio molecularorbital study of tetranitrogen tetrasulfide,N4S4[J].J Chem Res(S),1980(7):244-245.
[23] TURNER A G,MORTIMER F S.On the electronic structure of tetranitrogen tetrasulfide[J].Inorg Chem, 1966,5(5):906-910.
[24] FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al. All structures were optimized at the B3LYP/6-311++G (3df)level using Gaussian 98[M]∥Gaussian 98.Pittsburgh,PA:Gaussian,Inc,1998.
[25] BECKE A D.Density-functional thermochemistry.ⅲ. The role of exact exchange[J].J Chem Phys,1993,98 (7):5648-5652.
[26] LEE C,YANG W,PARR R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988, 37(2):785-789.
[27] WOLINSKI K,HILTON J F,PULAY P J.Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations[J].J Am Chem Soc,1990,112(23):8251-8260.
[28] SCHLEYER P V R,MAREKER C,DRANSFELD A,et al.Nucleus-independent chemical shifts:a simple and efficient aromaticity probe[J].J Am Chem Soc,1996,118 (26):6317-6318.
[29] SCHLEYER P V R,JIAO H,HOMMES N J R V E,et al.An evaluation of the aromaticity of inorganic rings:refined evidence from magnetic properties[J].J Am Chem Soc,1997,119(51):12669-12670.
[30] HERGES R,GEUENICH D.Delocalization of electrons in molecules[J].J Phys Chem A,2001,105(13): 3214-3220.
[31] FLUES W,SCHERER O J,WEISS J,et al.Crystal and molecular structure of the tetrasulfur pentanitride anion [J].Angew Chem Int Ed,1976,15(6):379-380.
[32] CHIVERS T,HILTS R W.Coordination chemistry of unsaturated cyclic and acyclic PNS and PNSe ligands [J].Coord Chem Rev,1994,137:201-232.
Through-space Homoconjugation and Homoaromaticity in S4N4and Se4N4
ZENG Hui,CHEN Xiumei,LÜXin*
(State Key Laboratory of Physical Chemistry of Solid Surface,Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China)
We show,by means of quantum chemical calculations,that two neutral inorganic compounds S4N4and Se4N4,which have long been well-characterized to have peculiar double-wedge cage structures,are homoaromatic.Their aromaticity can be mainly ascribed to the through-space homoconjugation between the separated(NXN)-subunits(X=S,Se).
bishomoaromaticity;through-space homoconjugation;electron delocalization;nucleus-independent chemical shift
O 64
A
0438-0479(2016)06-0793-04
10.6043/j.issn.0438-0479.201604035
2016-04-18 录用日期:2016-05-10
国家自然科学基金(21273177);高等学校博士学科点专项科研基金(20130121110004)
xinlu@xmu.edu.cn
曾慧,陈秀梅,吕鑫.S4N4和Se4N4中的跨空同共轭与同芳香性[J].厦门大学学报(自然科学版),2016,55(6): 793-796.
ZENG H,CHEN X M,LÜX.Through-space homoconjugation and homoaromaticity in S4N4and Se4N4[J].Journal of Xiamen University(Natural Science),2016,55(6):793-796.(in Chinese)
猜你喜欢
数学物理学报(2022年1期)2022-03-16 06:15:14
数学物理学报(2021年6期)2021-12-21 06:24:38
大学化学(2021年8期)2021-09-26 10:50:46
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
河北理科教学研究(2020年1期)2020-07-24 08:14:34
应用数学(2020年2期)2020-06-24 06:02:50
山东化工(2020年5期)2020-04-07 09:59:30
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
厦门大学学报(自然科学版)(2014年2期)2014-08-06 11:26:48
航天返回与遥感(2014年4期)2014-07-31 17:47:47
