
Meso位单取代卟啉及其配合物的合成、表征和光谱性质
2016-12-05 05:42:42沙秋月袁雪梅王小雨陈继超徐海军
无机化学学报 2016年7期
沙秋月 袁雪梅 王小雨 陈继超 徐 莉 徐海军*,
(1南京林业大学化学工程学院,江苏生物质能源与化学品重点实验室,南京210037)
(2南京林业大学现代分析测试中心,南京210037)
Meso位单取代卟啉及其配合物的合成、表征和光谱性质
沙秋月1袁雪梅1王小雨1陈继超1徐莉*,2徐海军*,1
(1南京林业大学化学工程学院,江苏生物质能源与化学品重点实验室,南京210037)
(2南京林业大学现代分析测试中心,南京210037)
设计合成了一系列新型的meso位N,N-二甲氨基苯基或N-苯基咔唑基单取代卟啉(5a~c)及其锌配合物(6a~c),用高分辨质谱、1H NMR、紫外-可见光谱及X射线单晶衍射方法等对结构进行了表征;研究了卟啉化合物及其配合物的热稳定性及荧光性质。结果表明,这些卟啉化合物及其锌配合物在400~410 nm之间具有强的吸收且具有很好的热稳定性,荧光量子产率在0.05~0.09;另外还分析了meso位不同取代基对光谱性质的影响。
卟啉;配合物;合成;晶体结构;荧光
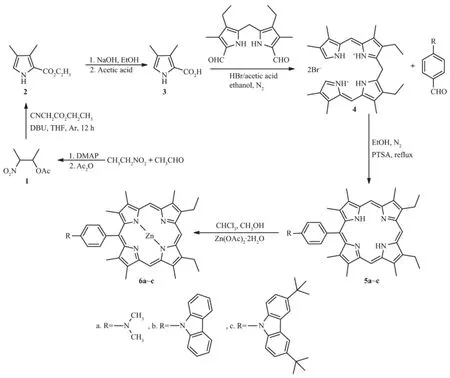
Scheme 1
1 Experimental
1.1Instruments
Column chromatography and TLC were performed on C-200(Wakogel)and Kieselgel 60 F254(Merck), respectively.1H NMR spectra were recorded on a Bruker DRX-600AVANCEⅢspectrometer.Chemical shifts for1H NMR spectra were expressed using CDCl3(δ=7.26)as the internal standard.The mass spectra data were obtained on a LTQ Orbitrap XL spectrometer in ESImode.UV-Vis spectra were carried out on a Shimadzu UV-3100 spectrophotometer.All the emission spectra were measured in FluoroLog-3-TCSPC(HORIBA Scientific,Edison)equipped with a 450 W CW xenon lamp and an Open-Electrode TECooled CCD Detector(Syncerity).Thermogravimetric analysis(TGA)measurements were performed on a Netzsch TG 209 analyzer under nitrogen at a scan rate of 10℃·min-1.
1.2M aterials
Unless otherwise noted,all chemicals and solvents were of analytical reagent grade and used directly as received.Dry dichloromethane was freshly distilled over CaH2under nitrogen.Dry tetrahydrofuran (THF)was distilled from sodium/benzophenone under nitrogen.2-Acetoxy-3-nitrobutane[25],3,3′-diethyl-5,5′-diformyl-4,4′-dimethyl-2,2′-dipyrrylmethane[26],4-(carbazol-9-yl)benzaldehyde[27]were prepared according to the related literature method,respectively.In addition,the introduction of tert-butyl group into the 3-and 6-positionsof carbazole could readily lead to the good solubility and 4-(3,6-di-tert-butyl-9H-carbazol-9-yl)-benzaldehyde was synthesized according to the similar procedure of 4-(carbazol-9-yl)benzaldehyde.
1.3Synthesis
The porphyrins and their complexes were synthesized as shown in Scheme 1.
1.3.1Synthesis of ethyl 3,4-dimethyl-1H-pyrrole-2-carboxylate(2)
Ethyl isocyanoacetate(7.6 mL,69.4 mmol)and 2-Acetoxy-3-nitrobutane(11.7 g,61.8 mmol)were dissolved in dry THF(160mL)underan Aratmosphere and cooled with an ice bath.DBU(21.6 mL,144.7 mmol)was slowly added and the solution was stirred. After dropping,the reaction mixture was stirred for another 12 h.Hydrochloric acid(1 mol·L-1)was added until the pH value of the solution became 7. The solution was diluted with water(50 mL).The organic materials were extracted with CHCl3(3×100 mL),and the combined organic layer was washed by water and brine,then dried with anhydrous sodium sulfate and the solvent was evaporated to get crude product which was further purified by column chromatography on silica gel with 60%(V/V)CH2Cl2-heptane as eluent.The white solid was obtained. Yield:8.4 g,81.1%.1H NMR(CDCl3):δ8.68(s,1 H), 6.66(s,1 H),4.31(q,2 H),2.27(s,3 H),2.01(s,3 H),1.35(t,3 H).
1.3.2Synthesis of 3,4-dimethyl-1H-pyrrole-2-carboxylic acid(3)
Compound 2(9.35 g,57.4 mmol)was dissolved in degassed absolute ethanol(150 mL)and the mixture was heated to reflux.A solution of sodium hydroxide(10.4 g,in 20 mL of water)was added dropwise and the reaction mixture was refluxed for 4 h.Themixture was then poured into ice,and diluted with water.After 10 min,acetic acid was added until pH=4.The solid was filtered using a sintered funnel and washed with water,dried in vacuo to afford compound 3(5.98 g,75%)as a white solid.1H NMR (DMSO-d6):δ10.96(s,1 H),6.64(s,1 H),2.15(s,3 H),1.91(s,3 H).
1.3.3Synthesis of 1,19-dideoxy-8,12-diethyl-2,3,7, 13,17,18-hexamethyl-a,c-biladiene dibromide (4)
Compound 3(3.91 g,28.10 mmol)and 3,3′-dicarboxy-5,5′-diformyl-4,4′-dimethyl-dipyrryl methane (4.02 g,14 mmol)were mixed and degassed ethanol (200 mL)was added and the mixture was heated to reflux for 10 min.Hydrobromic acid/acetic acid solution(33%,V/V,20mL)was added to the reaction mixture,which was checked by UV-Visible spectra, and the reaction was stopped after 5 min(the reaction should be stopped as soon as possible).The reaction mixture was cooled using dry ice bath,then added to cold diethyl ether(-20℃)(300 mL),followed byfiltration in sintered funnel and washing with cold ether.After drying in a desiccator,compound 4(6.50 g,77%)was acquired and used directly in the next step withoutany further purification.
1.3.4Synthesis of 5-(4-N,N-dimethylamino phenyl)-13,17-diethyl-2,3,7,8,12,18-hexamethylporphyrin(5a)
A solution of 4-(dimethylamino)benzaldehyde (149.1mg,1.0mmol)and compound 4(602.0 mg,1.0 mmol)in ethanol(150 mL)was heated to reflux,and nitrogen was bubbled through the system.Then,10.0 mL of a solution of para-toluene sulfonic acid(PTSA, 2.50 g)in ethanolwas slowly added during 18 h.The deep red solution was refluxed for 48 h under N2.The organic solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2,washed with saturated NaHCO3solution.The organic solution was separated by use of a separatory funnel.The solvent was removed under vacuum and the remaining residue was purified by repeated column chromatography on silica gelwith CH2Cl2-petroleum ether as eluents.The bright red band eluted was collected and dried. Recrystallization from CH2Cl2/methanol afforded compound 5a.Yield:(251.1 mg,44.1%).1H NMR (600 MHz,CDCl3):δ10.15(2H,s,meso-H),9.93(1H, s,meso-H),7.79(2H,m,Ar-H),7.04(2H,m,Ar-H), 4.04(4H,q,pyrro-C H2CH3),3.64(6H,s,pyrro-C H3), 3.53(6H,s,pyrro-C H3),3.22(6 H,s,-N(C H3)2),2.54(6 H,s,pyrro-C H3),1.88(6 H,t,CH2C H3),-3.16(1 H,s, N H),-3.27(1 H,s,N H);HRMS(ESI):Calcd.for C38H44N5(M+H+):570.359 1,Found:570.358 5;UV-Vis (CH2Cl2):λmax/nm(ε/(L·mol-1·cm-1)):403(2.143×105), 500(4.77×104),535(1.69×104),573(1.49×104),637 (1.28×104).
1.3.5Synthesis of 5-(4-N-carbazolylphenyl)-13,17-diethyl-2,3,7,8,12,18-hexamethyl-porphyrin (5b)
This compound was prepared in 31.5%yield (217.9mg),asdescribed for5a,starting from 4-(carbazol -9-yl)benzaldehyde(271.3mg,1mmol).1H NMR(600 MHz,CDCl3):δ10.20(2H,s,meso-H),9.98(1H,s, meso-H),8.29(4 H,m,Ar-H),7.93(2 H,d,Ar-H), 7.75(2 H,d,Ar-H),7.62(2 H,m,Ar-H),7.43(2 H,m, Ar-H),4.08(4 H,q,pyrro-C H2CH3),3.66(6 H,s, pyrro-C H3),3.59(6 H,s,pyrro-C H3),2.70(6 H,s, pyrro-C H3),1.89(6 H,t,CH2C H3),-3.14(1 H,s,N H), -3.28(1 H,s,N H);HRMS(ESI):Calcd.for C48H46N5(M+H+):692.374 8,Found:692.375 4;UV-Vis(CH2Cl2): λmax/nm(ε/(L·mol-1·cm-1)):293(4.38×104),403(2.937 ×105),500(1.99×104),535(1.04×104),570(1.34×104), 625(2.69×103).
1.3.6Synthesis of 5-(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)-13,17-diethyl-2,3,7,8,12, 18-hexamethyl-porphyrin(5c)
This compound was prepared in 32.9%yield (265 mg),as described for 5a,starting from 4-(3,6-ditert-butyl-9H-carbazol-9-yl)benzaldehyde(383.5 mg,1 mmol).1H NMR(600 MHz,CDCl3):δ10.19(2H,s, meso-H),9.97(1H,s,meso-H),8.30(2 H,s,Ar-H), 8.17(2 H,d,Ar-H),7.87(2 H,d,Ar-H),7.70(4 H,d, Ar-H),4.08(4 H,q,pyrro-C H2CH3),3.65(6 H,s, pyrro-C H3),3.57(6 H,s,pyrro-C H3),2.64(6 H,s,pyrro -C H3),1.90(6 H,t,CH2C H3),1.59(18 H,s,-C(C H3)3), -3.16(1 H,s,N H),-3.28(1 H,s,N H);HRMS(ESI): Calcd.forC56H62N5(M+H+):804.5000,Found:804.5013; UV-Vis(CH2Cl2):λmax/nm(ε/(L·mol-1·cm-1)):297(5.25 ×104),403(2.893×105),501(2.37×104),536(1.22×104), 570(1.85×104),622(5.53×103).
1.3.7Synthesis of zinc 5-(4-N,N-dimethylamino phenyl)-13,17-diethyl-2,3,7,8,12,18-hexamethyl -porphyrin(6a)
To a solution of porphyrin 5a(60 mg)in CHCl3(30mL)wasaddedwith asaturated solution of Zn(OAc)2·4H2O in MeOH(5 mL)and themixture was stirred overnight at room temperature.The reaction was checked by TLC.The reaction mixture was diluted with chloroform,washed with water two times,dried by MgSO4and concentrated to dryness in vacuo.The residue was purified by column chromatography(silica gel,50%(V/V)heptance-CH2Cl2)and recrystallized from CHCl3-methanol to afford the pure zinc complex 6a as a red solid(63 mg,Yield:95%).1H NMR(600 MHz,CDCl3):δ10.07(2 H,s,meso-H),9.91(1 H,s, meso-H),7.82(2 H,d,Ar-H),7.10(2 H,d,Ar-H),4.03 (4 H,q,pyrro-C H2CH3),3.59(6 H,s,pyrro-C H3),3.52 (6 H,s,pyrro-C H3),3.24(6 H,s,-N(C H3)2),2.55(6 H,s,pyrro-C H3),1.86(6 H,t,CH2C H3);HRMS(ESI):Calcd. for C38H42N5Zn(M+H+):632.272 6,Found:632.271 3; UV-Vis(CH2Cl2):λmax/nm(ε/(L·mol-1·cm-1)):405(1.632 ×105),534(1.22×105),570(1.08×105).
1.3.8Synthesis of zinc 5-(4-N-carbazolylphenyl)-13, 17-diethyl-2,3,7,8,12,18-hexamethyl-porphyrin (6b)
Compound 6b was prepared as a red solid in a similarway to the synthesis of 6a.Yield:98%,70mg.1H NMR(600 MHz,CDCl3):δ10.19(2H,s,meso-H), 10.08(1H,s,meso-H),8.32(2 H,d,Ar-H),8.29(2 H, d,Ar-H),7.94(2 H,d,Ar-H),7.77(2 H,d,Ar-H),7.62 (2 H,m,Ar-H),7.43(2H,m,Ar-H),4.12(4 H,q,pyrro-C H2CH3),3.66(6 H,s,pyrro-C H3),3.59(6 H,s,pyrro-C H3),2.71(6 H,s,pyrro-C H3),1.91(6 H,t,CH2C H3); HRMS(ESI):Calcd.for C48H43N5Zn(M+)·:753.281 0, Found:753.280 6;UV-Vis(CH2Cl2):λmax/nm(ε/(L· mol-1·cm-1):293(3.68×104),405(1.726×105),535 (1.05×104),573(8.71×103).
1.3.9Synthesis of zinc 5-(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)-13,17-diethyl-2,3,7,8,12, 18-hexamethyl-porphyrin(6c)
Compound 6c was prepared as a red solid quantitatively in a similar way to the synthesis of 6a.1H NMR(600 MHz,CDCl3):δ10.12(2H,s,meso-H), 9.98(1H,s,meso-H),8.29(2 H,s,Ar-H),8.24(2 H,m, Ar-H),7.92(2 H,d,Ar-H),7.72(2 H,d,Ar-H),7.69(2 H,m,Ar-H),4.06(4 H,q,pyrro-C H2CH3),3.62(6 H,s, pyrro-C H3),3.56(6 H,s,pyrro-C H3),2.66(6 H,s,pyrro -C H3),1.89(6 H,t,CH2C H3),1.57(18 H,s,-C(C H3)3); HRMS(ESI):Calcd.for C56H60N5Zn(M+):865.406 2, Found:865.406 4;UV-Vis(CH2Cl2):λmax/nm(ε/(L· mol-1·cm-1)):298(2.57×104),405(2.10×105),536(1.08× 104),571(1.23×104),633(1.81×103).
1.4Crystal structure determ ination
Single crystal diffraction data of 5a and 6a at 296 K was collected on a Bruker SMART APEXⅡCCD area-detector diffractometer using a graphitemonochromatized Mo Kαradiation(λ=0.071 073 nm). The structure was solved by direct methods and refined on F2by full-matrix least-squares techniques using SHELXL-97[28].All of the non-hydrogen atoms were refined by anisotropic thermal parameters,and hydrogen atoms bonded to the carbon and nitrogen atoms were generated geometrically and refined isotropically with the riding mode.The crystal data and structure refinementof 5a and 6a are summarized in Table1.
CCDC:1450675,5a;1471067,6a.
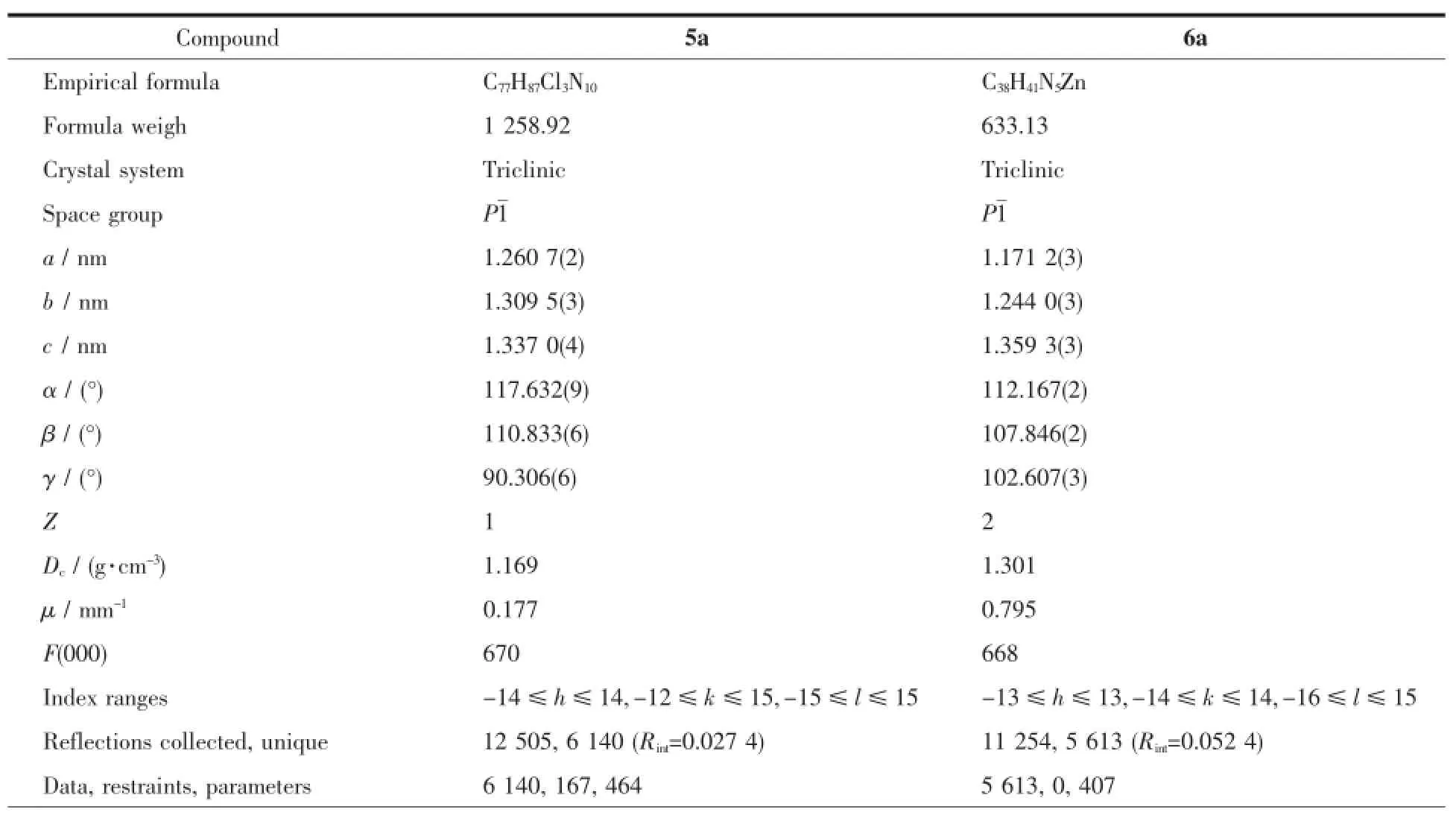
Table1 Crystal data and structure and refinement for compounds 5a and 6a

Continued Table1
2 Results and discussion
2.1Synthesis
The synthesis of meso-mono-substituted porphyrins 5a~c and theirmetal complexes 6a~c involved a multistep approach as shown in Scheme 1.Briefly,the porphyrin derivatives 5a~c were prepared by the condensation of 4 and different aromatic aldehyde in the presence of catalytic para-toluenesulfonic acid (PTSA)in higher yields of 44.1%,31.5%and 32.9%, respectively,compared with the convenient synthesis of the AB3-symmetric porphyrin[29].Their corresponding zinc complexes 6a~c were prepared in excellent yields(>90%)by starting from 5a~c and using Zn (OAc)2·2H2O in amethanol/chloroform mixture by use of the standard procedure.Their structures were characterized by1H NMR,ESI-HRMS and UV-Vis spectroscopy.
2.2Spectroscopic characterization
The proton NMR spectrum of 5a in CDCl3is shown in Fig.1.Peak assignments were made on the basis of chemical shifts,multiplicity,integrations,and spectral intercomparisons.As could be seen,signals atδ10.14 and 9.13 could be assigned to the meso protons.The two doublets atδ7.79 and 7.04 corresponded to the protons of phenyl ring.In the aliphatic region,the quadruplet atδ4.05~4.09 was attributed to the resonances of the-CH2protons.The signals observed atδ3.64,3.53 and 2.54 were attributed to the three types of methyl protons of theβposition of porphyrin respectively,and the signal of methyl protons which closest to the phenyl subunit lay relatively upfield due to the shielding effect of the phenyl rings.The chemical shift of themethyl protons of-N(CH3)2appeared atδ3.22.The triplet signals at δ1.87~1.90 were ascribed to the-CH2CH3methyl protons.In addition,the two singlets atδ-3.16 and -3.27 for two protons were assigned to the internal NH for the porphyrin ring.The structure was also supported by high resolution mass spectrometry, whichgave theexpected(M+H)+peak at m/z=570.358 5. Similarly,porphyrin derivatives 5b~c and their corresponding zinc complexes 6a~c were characterized and their spectra data were in accordance with their proposed structures.
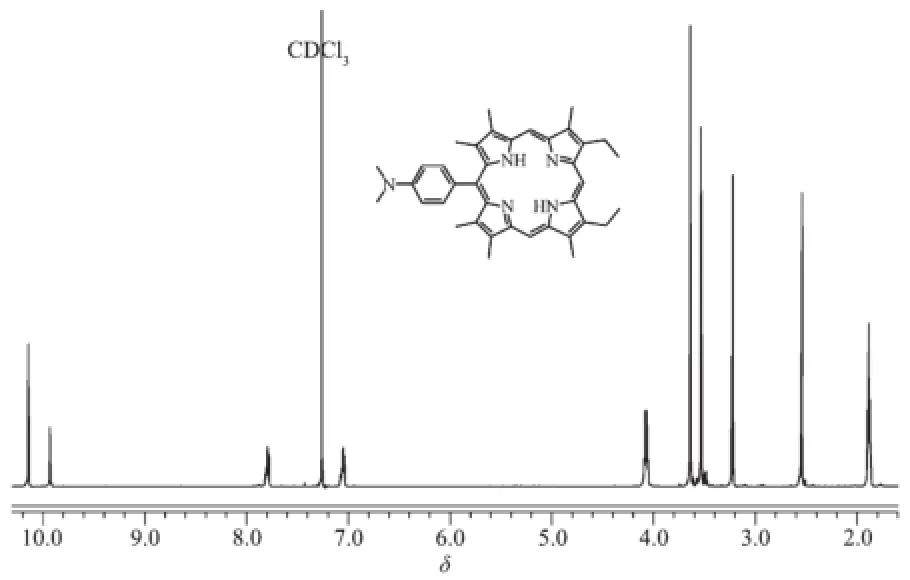
Fig.1 Partial1H NMR spectrum(600 MHz)of 5a in CDCl3
2.3X-ray crystal structure
X-ray crystallography analysis for 5a and 6a further supported the structure ofporphyrin derivatives. Single crystals of 5a and 6a suitable for X-ray crystallographic characterization were obtained by slow diffusion ofmethanol into the solution of 5a and 6a in chloroform.The compounds crystallized in the triclinic crystal system with space group P1.The resulting structures of 5a are shown in Fig.2,which confirms the asymmetric units consists of one porphyrinmolecule and half chloroform molecule(Fig. 2).The porphyrin structure exhibits a planar macrocycle and is very similar to that of previously reported porphyrins[30].The bond angles and bond distances are very similar to those found in the X-ray crystal structure of porphyrin derivatives[30].The substituted Cm position C1 exhibits Ca-Cm-Ca angle of 124.6(3)°,while the average Ca-Cm-Ca angles for C6,C11 and C16 is 128.9(3)°.With respect to theporphyrin(N4)plane,two ethyl groups are oriented above and below the N4 plane,respectively.The phenyl rings are almost orthogonal to the N4 plane and make an angle of 83.01(13)°due to the steric repulsion from the two methyl substituents at the pyrroleβ-positions(C…Ph 0.293 1~0.296 2 nm).

Fig.2 X-ray crystal structure of 5a(a)showing the side view(b)
The structure of the corresponding zinc porphyrin 6a is shown in Fig.3and exhibits very similar conformations and structural parameters.Its 24-atom porphyrin ring system is also nearly planar similar to 5a,exhibiting amean deviation of 0.006 87 nm and a maximum of 0.015 7 nm.N1…N3 distance is 0.407 5 nm and N2…N4 distance is 0.405 5 nm.The phenyl ring is virtually perpendicular to the porphyrin plane with a dihedral angle of 86.15°which is in agreement with the result observed in 5a.The zinc atom located on an inversion center with small mean plane deviations(0.007 11 nm).Zn-N bond distances are between 0.202 8 and 0.204 2 nm,which are similar to the reported zinc porphyrin derivatives[31].Two ethyl groups point to the same direction different from 5a. In addition,the plane defined by C1,N5 and C2 atoms is almost coplanar to the phenyl ring with a dihedral angle of 5.17(94)°.
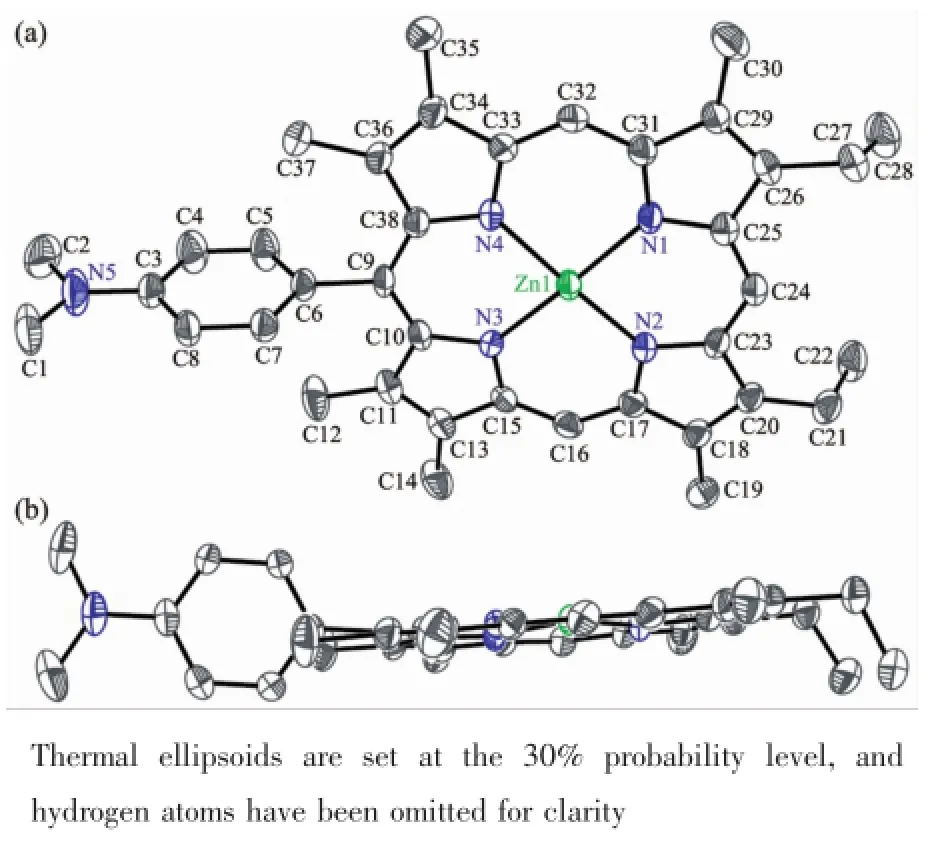
Fig.3 X-ray crystal structure of 6a(a)showing the side view(b)
2.4Absorp tion spectra
The absorption peaks of porphyrin derivatives 5a~c and 6a~c are presented in Table1.The UV-Vis spectra of free base porphyrins 5a~c are shown in Fig.4a and exhibit typical intense Soret band at 403 nm and four weaker Q bands at~500,535,570 and 637 nm,which are similar to that of meso-monosubstituted porphyrins such as 13,17-diethyl-2,3,7,8, 12,18-hexamethyl-5-phenylporphyrin[16]and 2-(13,17-diethyl-2,3,7,8,12,18-hexamethylporphyrin-5-yl)-5,5′,10,10′,15,15′-hexahexyltruxene[17].However,these bandsslightly blue-shiftcompared to the tetrakis(4-N,N -dimethylaminophenyl)porphyrin[32].Besides,the Soret and Q bands in porphyrins 5b and 5c are different from 5a with an additional absorption peak originating from the presence of carbazolyl units at~293 nm. Therefore,the absorption spectra of the two compounds 5b and 5c show the features of both porphyrin and carbazolyl subunits and suggest that there are no substantial interaction between the attached moiety and the porphyrin ring in the ground state.The electronic absorption spectra of the metalloporphyrins 6a~c are shown in Fig.4b.The compounds 6a~c show an intense absorption peak for the Soret bands at 405 nm and small absorption peaks for the Q-bands at~535 and~671 nm,which are almost equal to that of 13,17-diethyl-2,3,7,8,12,18-hexamethyl-5-(4-(diphenylamino)phenyl)porphyrinatozinc[18].Their Soret bands were found to red-shift by 2 nm in comparison to their corresponding free base porphyrin derivatives.In addition,comparing the absorption spectra data of 5a~c and 6a~c with thatof the literature[18],the results show that different meso-substituents of porphyrins have little impact on the electronic absorption region. The main reason could be the presence of the strong steric hindrance between theβ-methyl of the pyrrole ring and the meso aromatic rings which leaded to the lack of the substantial conjugation interaction between themeso aryl and the porphyrin ring.The results have also been proved by their crystal structures.

Table1 Electronic absorption and em ission data for the porphyrin derivatives 5a~c and 6a~c

Fig.4 Absorption spectra of porphyrins 5a~c(a)and 6a~c(b)in CH2Cl2
2.5Em ission spectroscopy
The fluorescence spectra of compounds 5a~c and 6a~c in 1%pyridine-toluene solution at excitation wavelength of 400 nm are shown in Fig.5.The porphyrins 5a~c show emission bands at~633 nm and a shoulder at~698 nm with almost equal intensities,and the porphyrins 6a~c show emission bands at~588 nm and a shoulder at~635 nm.Thefluorescence bands of 6a~c blue-shift by 45~63 nm with respect to their corresponding ligands 5a~c.On excitation at 400 nm,the fluorescence bands of all the compounds blue-shift by~20 nm in contrast to TPP and Zn-TPP,respectively.

Fig.5 Emission spectra of 5a~c(a)and 6a~c(b)in 1%pyridine-toluene solution
2.6Fluorescence quantum yields
The fluorescence quantum yields of these compounds were determined by comparing with a calibration standard of zinc phthalocyanine in toluene solution containing 1%pyridine presenting a fluorescencequantum yield of0.3.The Fluorescencequantum yields were calculated according to the following equation:
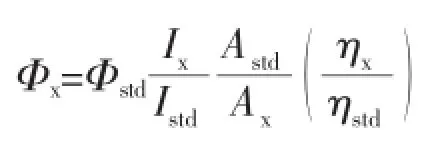
where the‘x’and‘std’subscripts refer to sample and standard(the reference),respectively,Φis the photoluminescence quantum yield,I is the integrated intensity of emission spectra,A stands for the absorbance at the excitation wavelength,andηis the refractive index of the solvent used in the measurement.
Values of quantum yields of the free base porphyrin derivatives and their metal complexes are reported in Table1.The quantum yields of 6a and 6c are slightly higher than their parent compound 5a and 5c,but6b isalmostequivalentto5b.Theseobservations may be explained by the non-conjugation interaction between the meso-aryl group and porphyrin ring. Further photophysical studies are currently under investigation.
2.7Thermal analysis
The thermal properties of the porphyrin derivatives have been investigated by TGA.The results are shown in Fig.6.Two stages of thermal decomposition were observed for 5a.The first began at 143℃and ended at 180℃with a small loss of 4%.The second stage of decomposition began at 350℃and continued up to a temperature of 700℃,at which complete decomposition of the material had not occurred.All other porphyrin derivatives only showed one major thermal events at approximately 403℃and complete decomposition had not been observed up to 700℃ similar to5a.Inaddition,thedecomposition temperature of zinc complexes 6a and 6c increases by approximately 10℃relative to their corresponding free base porphyrin 5a and 5c,indicating an increase in the thermal stability of the zinc complexes.These results show that all the porphyrin derivatives feature good thermal stability.
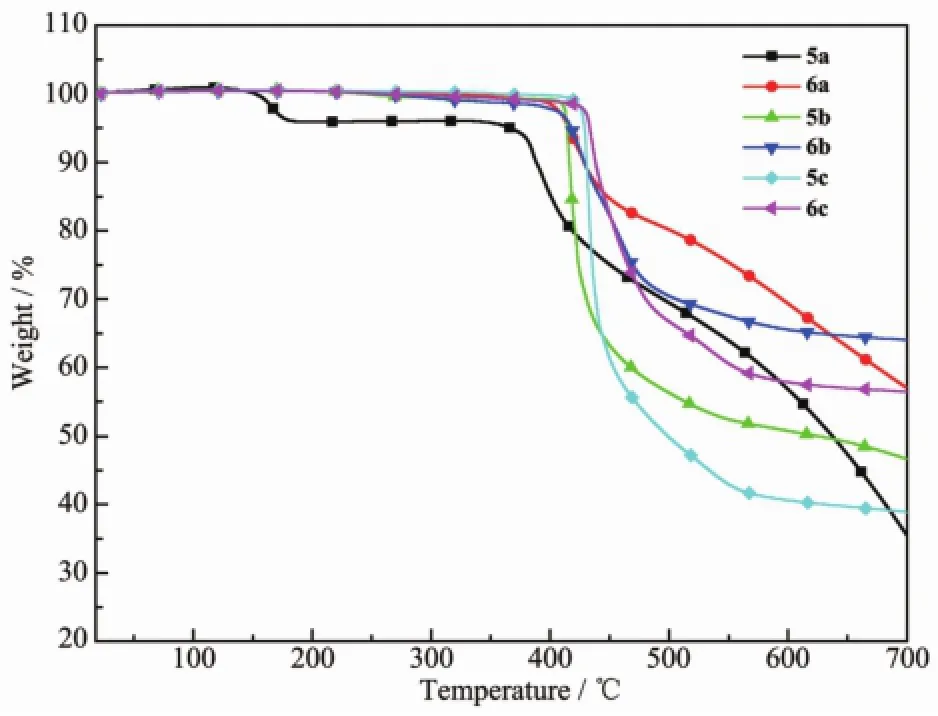
Fig.6 Thermogravimetric curves for 5a~c and 6a~c
3 Conclusions
In summary,three new meso-mono substituted porphyrin derivatives have been successfully synthesized and characterized.Their corresponding zinc complexes were prepared in high yield.These porphyrin derivatives can emit essentially red light after 400 nm irradiation.Different substituents of mesoposition have no remarkable effect on the spectra properties,which can be attributed to the possible steric hindrance between themeso phenyl substituents andβmethyl group.In addition,they present good thermal stability.Accordingly,these porphyrin derivatives might be promising candidates for luminescent materials.
Supporting information isavailableathttp://www.wjhxxb.cn
References:
[1]Jurow M,Schuckman A E,Batteas JD,et al.Coord.Chem. Rev.,2010,254(19/20):2297-2310
[2]Rangasamy S,Ju H,Um S,et al.J.Med.Chem.,2015,58 (17):68646874
[3]WANG Ling-Yun(汪凌云),CAO De-Rong(曹德榕).Chin.J.Org.Chem.(有机化学),2012,32(12):2248-2264
[4]REN Qi-Zhi(任奇志),LIU Shuang-Yan(刘双艳),WANG Ai-Qin(王爱琴),etal.Chinese J.Inorg.Chem.(无机化学学报), 2008,24(9):1438-1443
[5]Chan K,Guan X,Lo V,et al.Angew.Chem.Int.Ed.,2014, 53(11):2982-2987
[6]WU Di(吴迪),SHEN Zhen(沈珍),XUE Zhao-Li(薛兆历), et al.Chinese J.Inorg.Chem.(无机化学学报),2007,23(1): 1-14
[7]Rawson J,Stuart A C,You W,et al.J.Am.Chem.Soc., 2014,136(50):17561-17569
[8]Lu J,Li H,Liu S,et al.Phys.Chem.Chem.Phys.,2016,18 (9):6885-6892
[9]Arrechea S,Clifford J,PellejàL,etal.Dyes Pigm.,2016,126: 147-153
[10]Xu J,Wu J,Zong C,et al.Anal.Chem.,2013,85(6):3374-3379
[11]Hessenauer-Ilicheva N,Franke A,Meyer D,et al.Chem. Eur.J.,2009,15(12):2941-2959
[12]Cheng F,Wu X,Liu M,et al.Sens.Actuators B,2016,228: 673-678
[13]Liu X,Liu X,Tao M,et al.J.Mater.Chem.A,2015,3(25): 13254-13262
[14]Kral V,Kralova J,Kaplanek R,etal.Physiol.Res.,2006,55 (Suppl.2):S3-S26
[15]Mahmood M,Liu H,Wang H,etal.Tetrahedron Lett.,2013, 54(44):5853-5856
[16]Kolodina E,Syrbu S,Semeikin A,etal.Russ.J.Org.Chem., 2010,46(1):138-143
[17]Xu H,Du B,Gros C,et al.J.Porphyrins Phthalocyanines, 2013,17(1/2):44-55
[18]Takai A,Gros C,Barbe J,et al.Chem.Eur.J.,2011,17(12): 3420-3428
[19]Senge M,Kalisch W,Bichoff I.Chem.Eur.J.,2000,6(15): 2721-2738
[20]Lee M,Seo K,Song H,et al.Tetrahedron Lett.,2011,52 (30):3879-3882
[21]Nowak-Król A,Gryko D,Gryko D T.Chem.Asian J.,2010, 5(4):904-909
[22]Ormond A,Freeman H.Dyes Pigm.,2013,96:440-448
[21]Goldberg P,Pundsack T,Splan K.J.Phys.Chem.A,2011, 115(38):10452-10460
[22]Panda M,Sharma G,Thomas K,et al.J.Mater.Chem.,2012, 22(16):8092-8102
[23]Loiseau F,Campagna S,Hameurlaine A,et al.J.Am.Chem. Soc.,2005,127(32):11352-11363
[24]Xu T,Lu R,Liu X,et al.J.Org.Chem.,2008,73(5):1809-1817
[25]Matsuo T,Hayashi A,Abe M,et al.J.Am.Chem.Soc., 2009,131(42):15124-15125
[26]Juillard S,Simonneaux G.Synlett,2006(17):2818-2820
[27]Sun T,Pan Y,Wu J,et al.Transition Met.Chem.,2007,32 (4):449-455
[28]Sheldrick GM.Acta Crystallogr.A,2008,A64:112-122
[29]Ballatore M,Spesia M,Milanesio M,et al.Eur.J.Med. Chem.,2014,83:685-694
[30]Luguya R,Jaquinod L,Fronczek F,etal.Tetrahedron,2004, 60(12):2757-2763
[31]Byrn M,Curtis C,Hsiou Y,etal.J.Am.Chem.Soc.,1993, 115(21):9480-9497
[32]SUN Zhe(孙哲),JIA Li-Xia(贾立夏),LIYan-Rong(李艳荣), etal.Chin.J.Org.Chem.(有机化学),2010,30(4):546-550
Syntheses,Characterizations,Spectroscopic Properties of Meso-mono-substituted Porphyrins and Their Metal Com p lexes
SHA Qiu-Yue1YUAN Xue-Mei1WANG Xiao-Yu1CHEN Ji-Chao1XU Li*,2XU Hai-Jun*,1
(1College of Chemical Engineering,Jiangsu Key Lab of Biomass-based Green Fuels and Chemicals,Nanjing Forestry University,Nanjing 210037,China)
(2Advanced Analysis and Testing Center,Nanjing Forestry University,Nanjing 210037,China)
A series of novel asymmetricalmeso-mono-substituted porphyrin derivatives and their zinc complexes with N,N-dimethylphenylamine phenyland N-phenyl-carbazolewere synthesized.Their structuresand photophysical properties were characterized by MS,1H NMR,single Crystal X-ray diffraction analysis,thermogravimetric analysis(TGA),UV-Vis absorption spectroscopy,and photoluminescence spectroscopy.The results showed that free base porphyrins(5a~c)and their zinc porphyrins(6a~c)exhibited a strong absorption in the range of 400~450 nm with molar absorptivity of~105L·mol-1·cm-1.Their emission were characterized in solution by large Stokes shifts(80~290 nm)and the fluorescence quantum yields ranged from 0.05 to 0.09.The compounds had also shown high thermal stability.In addition,the effect of the different substituent groups of themeso positions on the photophysical propertieswas studied.CCDC:1450675,5a;1471067,6a.
porphyrins;complex;synthesis;crystal structure;photoluminescence
0 Introduction
Porphyrin and porphyrin derivatives have great attention and study formany years due to theirunique chemicaland opticalproperties,such as good photostability,rigidmolecular structuresandmany reaction sites.They play important roles inmany fields and show a wide range of perspective applications in molecularelectronic devices[1],photodynamic therapy[2-3], catalytic reactions[4-5],solar energy transduction[6-9], mimicking enzymes[10-11],and sensors[12-13].The diversity in functions has a great impetus on the extensive development of the design and synthesis of novel porphyrin derivatives.Among meso-substituted porphyrins,symmetrical porphyrins are usually more easily synthesized than unsymmetrical porphyrins[14-15]. However,to the best of our knowledge,only a few of meso-mono-substituted porphyrin derivatives have been synthesized[16-19].The nature and propensity of meso-substituents aremainly responsible for tuning the electronic,chemicaland thermalpropertiesof porphyrin derivatives[20-22].N,N-dimethylphenylamine phenyl and N-phenyl-carbazole groups with the strong electron ability by introducing the meso position of porphyrin ring can be used for tuning their optical,electrochemical,and photophysical properties[21-24].However, meso-mono-substituted porphyrin derivatives with N, N-dimethylphenylamine phenyl or N-phenyl-carbazole groups could not be characterized up to now probably due to the poor reactivity of 4-(dimethylamino)benzaldehyde or 4-(carbazol-9-yl)benzaldehyde under normal Lindsey porphyrin condensation.Therefore,we herein report the synthesis,characterization,and spectroscopic property studies of a series of new mono-substituted porphyrin derivatives(5a~c)and their zinc complexes (6a~c)with N,N-dimethylphenylamine phenyl or N-phenyl-carbazole groups(Scheme 1).
O614.24+1
A
1001-4861(2016)07-1293-10
10.11862/CJIC.2016.166
2016-04-11。收修改稿日期:2016-05-31。
国家自然科学基金(No.21301092)、霍英东教育基金会基金(No.141030)、江苏省生物质绿色燃料与化学品重点实验室开放基金(No.JSBGFC12002)、江苏高校优势学科建设工程项目、青蓝工程及江苏省高等学校大学生创新创业训练计划项目(No.201410298009z)资助。
*通信联系人。E-mail:xuhaijun_jx@163.com;xuliqby@163.com;会员登记号:S06N8692M1305。
猜你喜欢
南京林业大学学报(自然科学版)(2023年1期)2023-03-30 03:40:00
南京林业大学学报(自然科学版)(2022年3期)2022-11-30 01:46:54
南京林业大学学报(自然科学版)(2022年5期)2022-10-19 02:07:38
南京林业大学学报(自然科学版)(2021年5期)2021-10-10 06:27:44
分析测试技术与仪器(2019年3期)2019-10-09 10:16:30
劳动保护(2019年3期)2019-05-16 02:38:16
西南交通大学学报(2018年6期)2018-12-18 02:22:26
现代园艺(2018年1期)2018-03-15 07:56:41
安全(2015年6期)2016-01-19 06:19:31
中国科技信息(2015年17期)2015-11-02 12:48:29
