
Methylprednisolone exerts neuroprotective effects by regulating autophagy and apoptosis
2016-12-02 07:05:41WeiGaoShuruiChenMengyaoWuKaiGaoYuanlongLiHongyuWangChenyuanLiHongLi
中国神经再生研究(英文版) 2016年5期
Wei Gao, Shu-rui Chen, Meng-yao Wu, Kai Gao, Yuan-long Li, Hong-yu Wang, Chen-yuan Li, Hong Li
Department of Biochemistry, Liaoning Medical University, Jinzhou, Liaoning Province, China
RESEARCH ARTICLE
Methylprednisolone exerts neuroprotective effects by regulating autophagy and apoptosis
Wei Gao#, Shu-rui Chen#, Meng-yao Wu, Kai Gao, Yuan-long Li, Hong-yu Wang, Chen-yuan Li, Hong Li*
Department of Biochemistry, Liaoning Medical University, Jinzhou, Liaoning Province, China
Graphical Abstract
#These authors contributed equally to this study.
orcid: 0000-0002-5446-6717
(Hong Li)
Methylprednisolone markedly reduces autophagy and apoptosis after secondary spinal cord injury. Here, we investigated whether pretreatment of cells with methylprednisolone would protect neuron-like cells from subsequent oxidative damage via suppression of autophagy and apoptosis. Cultured N2a cells were pretreated with 10 µM methylprednisolone for 30 minutes, then exposed to 100 µM H2O2for 24 hours. Inverted phase contrast microscope images, MTT assay, flow cytometry and western blot results showed that, compared to cells exposed to 100 µM H2O2alone, cells pretreated with methylprednisolone had a significantly lower percentage of apoptotic cells, maintained a healthy morphology, and showed downregulation of autophagic protein light chain 3B and Beclin-1 protein expression. These findings indicate that methylprednisolone exerted neuroprotective effects against oxidative damage by suppressing autophagy and apoptosis.
nerve regeneration; spinal cord injury; methylprednisolone; oxidative stress; N2a cells; autophagy; light chain 3B; Beclin-1; apoptosis; neuroprotection; H2O2; neural regeneration
Introduction
The pathophysiology of spinal cord injury (SCI) involves primary and secondary mechanisms (Silva et al., 2014; Gao et al., 2015b). Secondary SCI is caused by a series of biochemical events triggered by the primary injury. The pathology of secondary SCI involves microcirculatory changes, oxidative stress, ischemia, apoptosis, necrosis, and edema. These secondary effects further aggravate the primary injury and cause destructive lesions in the region surrounding the injury site (Webb et al., 2010; Gao et al., 2015a). Following acute SCI, an increase in free radical production leads to lipid peroxidation damage; decreasing oxidative stress would therefore greatly reduce secondary injury following trauma (Juurlink and Paterson, 1998).
Methylprednisolone (MP), a synthetic glucocorticoid hormone, is the most commonly used anti-inflammatory and antioxidant drug in the treatment of acute SCI (Lee et al., 2008; Bains and Hall, 2012). However, the effects of MP on SCI have been questioned in recent studies, and the mechanisms underlying its preventive effects on secondary pathological damage after SCI remain poorly understood (Liu et al., 2009; Mazzocca et al., 2013; Fehlings et al., 2014;Harrop, 2014). Following SCI, MP inhibits inflammation and promotes recovery of neurological function. To date, the main focus of research into the neuroprotective effect of MP has been from the aspect of glucocorticoid receptor and anti-inflammatory mechanisms, and studies of its antioxidant mechanism remain scarce (Xu et al., 1998; Mazzocca et al., 2013; Boyaci et al., 2014).
MP has been shown to regulate autophagy, but findings have been inconsistent. In a study using osteoblasts, application of MP markedly increased autophagic activity (Yao et al., 2015). However, Chen et al. (2012) demonstrated that MP suppressed autophagy. Furthermore, it remains unclear whether MP exerts its neuroprotective effect by reducing light chain 3B (LC3B) and Beclin-1 expression. LC3B and Beclin-1 are two key markers for autophagy. Apoptosis and autophagy are extensive following secondary SCI. Autophagy might be induced and activated by primary SCI (Kanno et al., 2009a, b, 2011; Walker et al., 2012; Hou et al., 2014). However, several studies have shown that apoptosis is also an important cell death mechanism after SCI (Crowe et al., 1997; Springer et al., 1999), and Lee et al. (2008) confirmed that MP selectively inhibits microglial apoptosis, which may be associated with its neuroprotective effect.
Here, we established an in vitro model of oxidative damage in N2a cells, using H2O2, and explored the effects of MP on autophagy and apoptosis after exposure to oxidative stress.
Materials and Methods
Cell culture
Frozen mouse neuroblastoma cells (N2a cells; Guangzhou Jiniou Co., Ltd., Guangzhou, China) were resuscitated in a water bath at 37°C for 3 minutes, placed in a 15 mL centrifuge tube, incubated with Dulbecco's modified Eagle's medium/F12 containing 10% fetal bovine serum (twice the volume of frozen liquid) at room temperature, and centrifuged. After removal of the supernatant, cells were incubated with Dulbecco's modified Eagle's medium (DMEM)/F12 containing 10% fetal bovine serum, at 37°C, 5% CO2and saturated humidity. Cells were passaged at approximately 80% confluence, and collected for use at passages 3—6. The medium was replaced every other day.
3-(4,5-Cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell proliferation rate assay
Logarithmic-phase N2a cells were seeded on a 96-well plate at 1×105cells per well in 200 µL cell suspension. Phosphate buffered saline (PBS) was added to the surrounding wells. The plate was incubated at 37°C and 5% CO2for 24 hours, to allow the cells to adhere. The cells were then allocated to four groups: cells in the control group were incubated in DMEM containing 10% fetal bovine serum; in the H2O2group, 0, 20, 40, 60, 80, 100, 120, 140, 160 or 200 µM H2O2(Bori Site Trading Co., Ltd., Shenyang, China) was added to the culture medium; cells in the MP group were treated with 0, 0.5, 1, 2, 5, 10, 20, 50, 100 or 200 µM MP (Melonepharma, Dalian, China); and in the H2O2+ MP group, cells were pretreated with 0.1, 1, 5, 10, 50 or 100 µM MP for 30 minutes, then 100 µM H2O2was added. Each group contained four parallel wells. Cells were observed 24 hours later under an inverted phase contrast microscope (Olympus, Tokyo, Japan). Subsequently, 20 µL MTT (Sigma, St. Louis, MO, USA) was added to each well for 4 hours. The medium was removed and the cells were incubated with 150 µL of dimethyl sulfoxide for 10 minutes at 37°C. Optical density (OD) values were measured at 570 nm with a microplate reader (Bio-Rad, Hercules, CA, USA). Cell proliferation rate was calculated as
Cell counting and morphology
Cells were seeded in the culture dish and the medium was replaced every 3—4 days for approximately 1 week. After digestion with trypsin (Gibco, Carlsbad, CA, USA), cells were incubated in six-well plates for 24 hours. Cells were allocated to three groups: in the H2O2group, cells were exposed to 100 µM H2O2for 24 hours; cells in the H2O2+ MP group were pretreated with 10 µM MP for 30 minutes, before exposure to 100 µM H2O2; control cells were in DMEM containing 10% fetal bovine serum. Cell morphology was observed at 24 hours under an inverted phase contrast microscope (Olympus).
Flow cytometry
Cells were incubated in 6-well plates for 24 hours, then grouped and treated as described above (see Cell counting and morphology). A single-cell suspension was made using trypsin without ethylenediamine tetraacetic acid, and centrifuged at 300 × g for 3 minutes. Following removal of the supernatant, cells were washed twice with precooled PBS, and centrifuged in 1 mL annexin V (Tianjin Sungene Biotech Co., Ltd., Tianjin, China) for 10 minutes. Cells were adjusted to 106/mL. Cell suspension was centrifuged and washed three times with PBS. Samples (100 µL) were added to Eppendorf tubes with 5 µL annexin V-APC (Tianjin Sungene Biotech Co., Ltd.) and 7-AAD (Tianjin Sungene Biotech Co., Ltd.), and mixed. The volume was made up to 500 µL with PBS and the tubes were incubated at room temperature for 15 minutes in the dark. Apoptosis was quantified by flow cytometry (BD FACSCanto II, BD Becton Dickinson, San Jose, CA, USA). Cell apoptosis rate was calculated as follows: number of apoptotic cells/total number of cells × 100%. Experiments were conducted twice.
Immunofluorescence staining
Cells were plated onto 24-well plates for 24 hours, then the medium was discarded and the cells were grouped and treated as described above (see Cell counting and morphology). The cells were then fixed in 4% paraformaldehyde at 4°C overnight. The next day, samples were washed three times with PBS, for 5 minutes each time. Triton X-100 (1%) was added for 15 minutes, and the samples were blocked with goat serum for 1 hour at 4°C. Samples were then incubated overnight with rabbit polyclonal LC3B antibody (1:500 dilution in goat serum; Abcam, Cambridge, MA, USA). The following day, the samples were washed with PBS as before,and incubated with goat anti-rabbit IgG/Cy3 in the dark for 2 hours at room temperature, followed by three 5-minute washes with PBS. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole for 15 minutes, followed by three more 5-minute washes with PBS. Protein localization was observed under a fluorescence microscope (Olympus).
Western blot assay
Cells were assigned to control, H2O2and H2O2+ MP groups as described above. After lysis, protein concentrations were measured using the bicinchoninic acid method. Lysates were electrophoresed on 5% stacking gel at 90 V, and 10% separating gel at 120 V, then wet-transferred onto polyvinylidene fluoride membranes at 350 mA for 1 hour 35 minutes, after immersing the dry membranes in methanol for 30 seconds and transfer buffer for 10 minutes. Membranes were blocked with 1% bovine serum albumin for 2 hours, and incubated with rabbit anti-LC3B polyclonal antibody (1:1,000; Cell Signaling, China), mouse anti-β-actin polyclonal antibody (1:1,000; Abcam), and rabbit anti-Beclin-1 polyclonal antibody (1:1,000; Abcam) at 4°C overnight. The next day, the membranes were rinsed in Tris-buffered saline with Tween-20 (TBST), three times for 10 minutes each time, incubated with goat anti-rabbit or anti-mouse secondary antibody (1:1,000; Bioss, Beijing, China) at room temperature for 2 hours, then washed with TBST as before. Proteins were visualized using enhanced chemiluminescence. Gray values of the protein bands (target protein/β-actin) were calculated using ImageJ2x software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± SD, and were analyzed using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Groups were compared using one-way analysis of variance and the least significant difference test. P< 0.05 was considered statistically significant.
Results
MP prevented H2O2-induced reduction in cell survival
N2a cells were exposed to different concentrations of H2O2for 24 hours. MTT assay showed that cell survival rate decreased with increasing concentrations of H2O2. At 100 µM H2O2, cell viability was approximately 50% of the control group value (0 µM H2O2; P < 0.001; Figure 1A).
To exclude the effects of MP on cell viability, N2a cells were exposed to different concentrations of MP for 24 hours. MP had no observable effect on cell viability (Figure 1B), confirming that it neither improved cell survival nor was cytotoxic.
After pretreatment of N2a cells with different concentrations of MP for 30 minutes, followed by 100 µM H2O2for 24 hours, MP markedly suppressed the reduction in cell viability induced by H2O2(P < 0.001; Figure 1C).
MP prevented H2O2-induced damage to cell morphology and number
Control cells were plump, with long dendrites and axons, indicating good growth. After exposure to H2O2, cell bodies floated and appeared round, and some processes retracted and were noticeably damaged, indicating that H2O2was cytotoxic. In the H2O2+ MP group there were significantly fewer damaged and floating cells than in the H2O2group (Figure 2).
MP prevented H2O2-induced apoptosis
Flow cytometry showed that there were significantly more apoptotic cells in the H2O2group than in the control group (P < 0.001), but fewer in the MP + H2O2group than in the H2O2group (P < 0.01; Figure 3).
MP prevented H2O2-induced upregulation of LC3B and Beclin-1
Immunofluorescence staining was used to detect LC3B expression and localization. LC3B was mainly expressed in the cytoplasm. Western blot assay revealed that expression levels of LC3B and Beclin-1 protein were significantly higher in the H2O2group than in the control group (P < 0.01), and significantly lower in the H2O2+ MP group than in the H2O2group (P < 0.05 or P < 0.001; Figure 4).
Discussion
Cytosolic LC3A fractions convert to autophagic membrane-form (phosphatidyl ethanolamine-conjugated) LC3B. When autophagy occurs, LC3B expression increases, and LC3B becomes selectively incorporated into the autophagic membrane. Therefore, LC3B expression is directly proportional to the number of autophagosomes, making LC3B a unique autophagosomal marker. The gene that encodes Beclin-1 is the first gene involved in a series of events that activate autophagy in mammals, and as such is a key marker of autophagy initiation (Walker et al., 2012; Fitzwalter and Thorburn, 2015). Cells undergoing autophagy, a self-degradative process, deliver cytoplasmic material and organelles to lysosomes for degradation (Magraoui et al., 2015). Apoptosis is another cell death mechanism, also under genetic control (Edinger and Thompson, 2004). Initial studies suggested that there were considerable differences between autophagy and apoptosis in terms of morphology, biochemistry, and molecular mechanism, but recent evidence indicates that the two processes can occur successively or simultaneously in the same cell (Marino et al., 2014; Mukhopadhyay et al., 2014; Fitzwalter and Thorburn, 2015). Our flow cytometry results showed that the proportion of apoptotic cells was markedly lower in the H2O2+ MP group than in the group exposed to H2O2alone. Furthermore, the immunofluorescence and western blot experiments showed that MP prevented the H2O2-induced upregulation of Beclin-1 and LC3B protein expression.
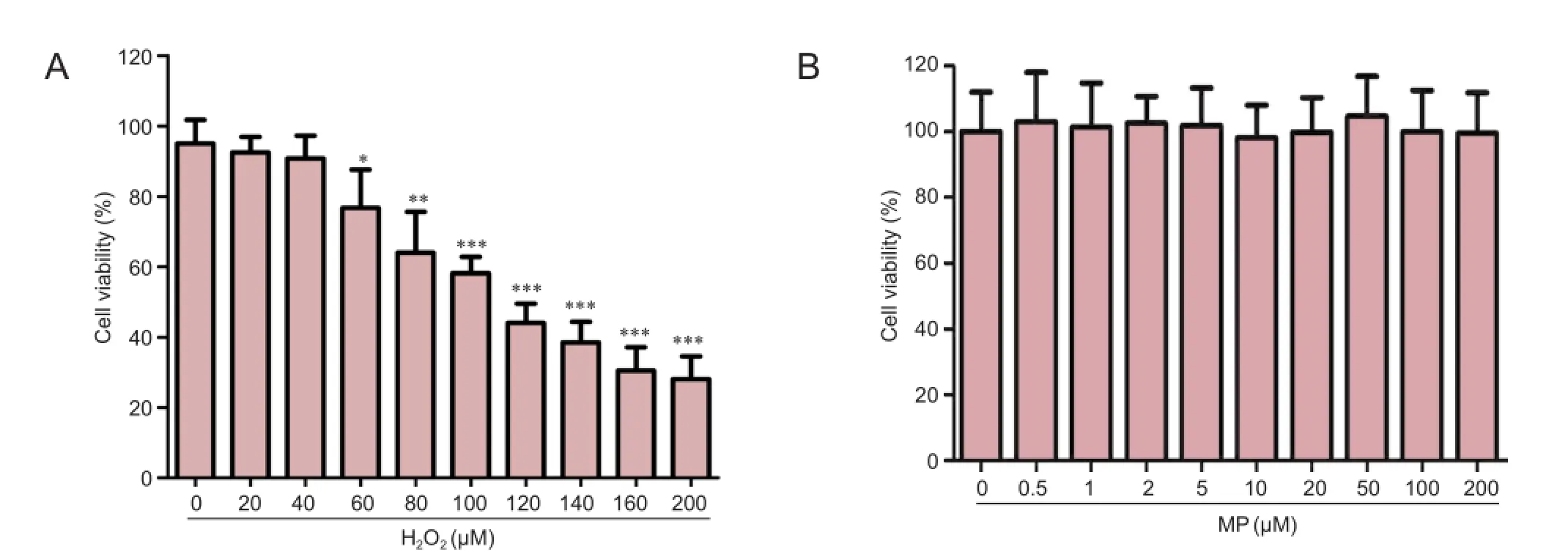

Figure 1 Neuroprotective effect of MP against oxidative damage induced by H2O2(100 μM) in N2a cells.

Figure 2 Effects of 10 μM MP on the morphology and number of N2a cells (inverted phase contrast microscope, × 400).
Our results clearly show that H2O2is cytotoxic in N2a cells, and autophagy and apoptosis were observed. This result supports the notion that autophagy and apoptosis exist simultaneously and interact with each other during periods of oxidative damage. However, pretreatment with MP decreased the rate of apoptosis, and inhibited autophagy, in N2a cells subsequently exposed to H2O2-induced oxidative stress. We also investigated whether the neuroprotective effect of MP depended on the downregulation of key proteins in the autophagy and apoptosis signaling pathways. Our results show that MP pretreatment diminished oxidative stress-induced cell damage and protected cells via inhibition of autophagy and apoptosis.
In conclusion, MP has anti-apoptotic and anti-autophagic effects in neuron-like cells exposed to oxidative damage. We are the first to show that MP downregulates LC3B and Beclin-1 protein expression. However, this is only a preliminary investigation of the effects of MP on autophagic and apoptotic signaling pathways, and further experiments are needed to determine whether our results can be extrapolated to the whole organism. Nevertheless, the present results provide strong and novel evidence elucidating the neuroprotective mechanism of MP against oxidization.
Acknowledgments: We are very grateful to Xi-fan Mei from the Department of Orthopedics, the First Affiliated Hospital of Liaoning Medical University of China for selfless guidance and assistance. We would like to thank Jing Bi and Dong-he Han from the Liaoning Province Key Laboratory for Neurodegenerative Diseases of China for their valuable advice and technical support.
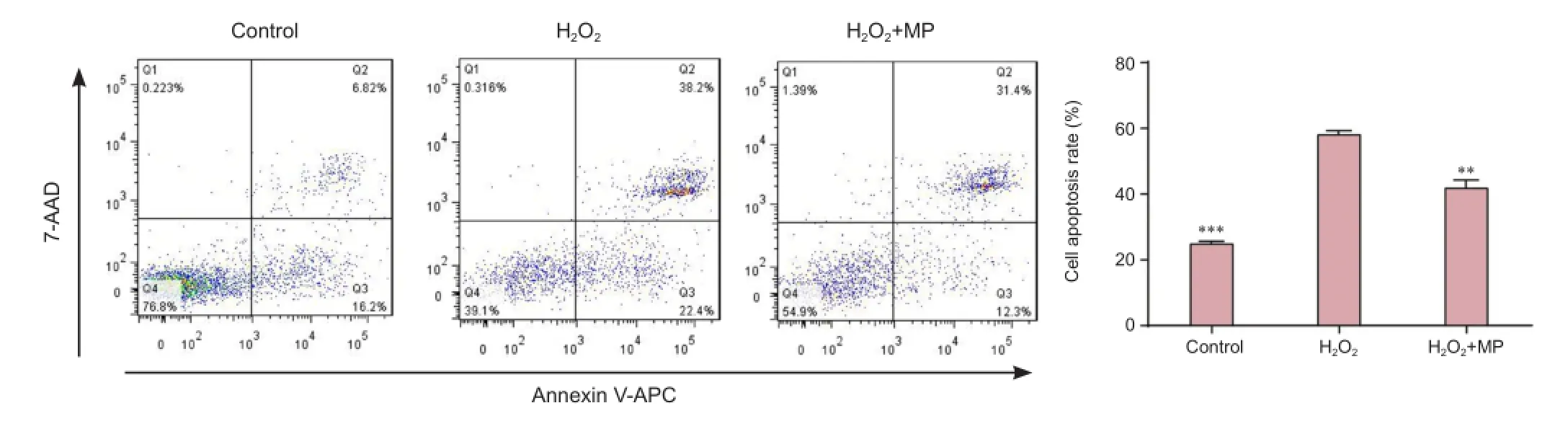
Figure 3 Pretreatment with MP protected against H2O2-induced apoptosis (flow cytometry).
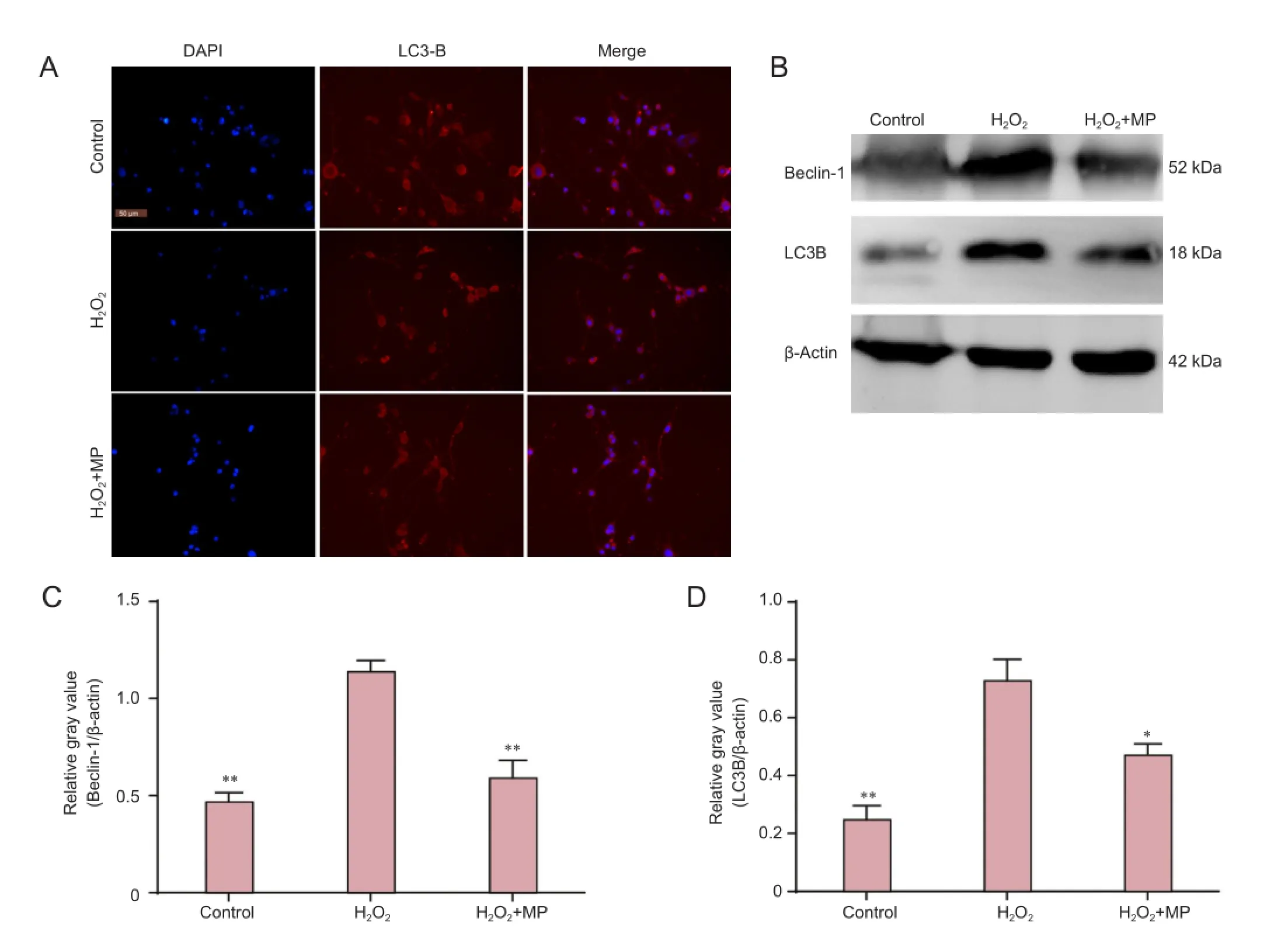
Figure 4 Pretreatment with MP protected against H2O2-induced LC3B and Beclin-1 protein upregulation in N2a cells.
Author contributions: WG conceived and designed the study, participated in cell culture, MTT, flow cytometry, immunofluorescence and western blot assay, analyzed data and wrote the paper. SRC provided data support, retrieved references, and ensured the integrity of the data. MYW participated in cell cul-ture, passage, resuscitation and cryopreservation. KG analyzed data. YLL participated in flow cytometry. HYW provided reference data and technical support. CYL obtained the funding, conceived and designed the study. HL served as a principle investigator. All authors approved the final version of the paper.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Bains M, Hall ED (2012) Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta 1822:675-684.
Boyaci MG, Eser O, Kocogullari CU, Karavelioglu E, Tokyol C, Can Y (2014) Neuroprotective effect of alpha-lipoic acid and methylprednisolone on the spinal cord ischemia/reperfusion injury in rabbits. Br J Neurosurg: 1-6.
Chen HC, Fong TH, Lee AW, Chiu WT (2012) Autophagy is activated in injured neurons and inhibited by methylprednisolone after experimental spinal cord injury. Spine (Phila Pa 1976) 37:470-475.
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3:73-76.
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16:663-669.
Fehlings MG, Wilson JR, Cho N (2014) Methylprednisolone for the treatment of acute spinal cord injury: counterpoint. Neurosurgery 61 Suppl 1:36-42.
Fitzwalter BE, Thorburn A (2015) Recent insights into cell death and autophagy. FEBS J 282:4279-4288.
Gao K, Shen Z, Yuan Y, Han D, Song C, Guo Y, Mei X (2015a) Simvastatin inhibits neural cell apoptosis and promotes locomotor recovery via activation of Wnt/beta-catenin signaling pathway after spinal cord injury. J Neurochem doi: 10.1111/jnc.13382.
Gao K, Wang YS, Yuan YJ, Wan ZH, Yao TC, Li HH, Tang PF, Mei XF (2015b) Neuroprotective effect of rapamycin on spinal cord injury via activation of the Wnt/beta-catenin signaling pathway. Neural Regen Res 10:951-957.
Harrop JS (2014) Spinal cord injury: debating the efficacy of methylprednisolone. Neurosurgery 61 Suppl 1:30-31.
Hou H, Zhang L, Liu D, Mao Z, Du H, Tang P (2014) Acute spinal cord injury in rats induces autophagy activation. Turk Neurosurg 24:369-373.
Juurlink BH, Paterson PG (1998) Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med 21:309-334.
Kanno H, Ozawa H, Sekiguchi A, Itoi E (2009a) The role of autophagy in spinal cord injury. Autophagy 5:390-392.
Kanno H, Ozawa H, Sekiguchi A, Itoi E (2009b) Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol Dis 33:143-148.
Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E (2011) Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine (Phila Pa 1976) 36:E1427-1434.
Lee JM, Yan P, Xiao Q, Chen S, Lee KY, Hsu CY, Xu J (2008) Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury. J Neurosci 28:3141-3149.
Liu JC, Patel A, Vaccaro AR, Lammertse DP, Chen D (2009) Methylprednisolone after traumatic spinal cord injury: yes or no? PM R 1:669-673.
Magraoui FE, Reidick C, Meyer HE, Platta HW (2015) Autophagy-related deubiquitinating enzymes involved in health and disease. Cells 4:596-621.
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81-94.
Mazzocca AD, McCarthy MB, Intravia J, Beitzel K, Apostolakos J, Cote MP, Bradley J, Arciero RA (2013) An in vitro evaluation of the anti-inflammatory effects of platelet-rich plasma, ketorolac, and methylprednisolone. Arthroscopy 29:675-683.
Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK (2014) Autophagy and apoptosis: where do they meet? Apoptosis 19:555-566.
Silva NA, Sousa N, Reis RL, Salgado AJ (2014) From basics to clinical: a comprehensive review on spinal cord injury. Prog Neurobiol 114:25-57.
Springer JE, Azbill RD, Knapp PE (1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 5:943-946.
Walker CL, Walker MJ, Liu NK, Risberg EC, Gao X, Chen J, Xu XM (2012) Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS One 7:e30012.
Webb AA, Ngan S, Fowler JD (2010) Spinal cord injury I: a synopsis of the basic science. Can Vet J 51:485-492.
Xu J, Fan G, Chen S, Wu Y, Xu XM, Hsu CY (1998) Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res 59:135-142.
Yao W, Dai W, Jiang L, Lay EY, Zhong Z, Ritchie RO, Li X, Ke H, Lane NE (2015) Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporos Int 27:283-294.
Copyedited by Slone-Murphy J, Robens J, Wang J, Qiu Y, Li CH, Song LP, Zhao M
10.4103/1673-5374.182711 http://www.nrronline.org/
How to cite this article: Gao W, Chen SR, Wu MY, Gao K, Li YL, Wang HY, Li CY, Li H (2016) Methylprednisolone exerts neuroprotective effects by regulating autophagy and apoptosis. Neural Regen Res 11(5)∶823-828.
Funding: This research was supported by the National Natural Science Foundation of China, No. 81171799, 81471854; the Science and Technology Research Project of Education Department of Liaoning Province of China, No. L2013333; the “College Students' Science and Technology Innovation Project” of Liaoning Medical University Principal Fund-Aohong Boze Fund of China, No. 2014D08; the Liaoning Medical University Principal Fund-Aohong Boze Graduate Student Science Research Innovation Fund, No. AH2014017.
Accepted: 2015-12-22
*Correspondence to: Hong Li, M.D., jzmu_lihong@sina.com.
- 中国神经再生研究(英文版)的其它文章
- Recovery of injured fornical crura following neurosurgical operation of a brain tumor: a case report
- Gender difference in the neuroprotective effect of rat bone marrow mesenchymal cells against hypoxiainduced apoptosis of retinal ganglion cells
- Vitamin B complex and vitamin B12levels after peripheral nerve injury
- Methylprednisolone microsphere sustained-release membrane inhibits scar formation at the site of peripheral nerve lesion
- A self-made, low-cost infrared system for evaluating the sciatic functional index in mice
- Repetitive magnetic stimulation affects the microenvironment of nerve regeneration and evoked potentials after spinal cord injury