
离子液体表面活性剂在水溶液中的自组装及其调控研究进展
2016-11-18 07:28王慧勇李虹培崔国凯李志勇王键吉
物理化学学报 2016年1期
王慧勇李虹培崔国凯李志勇王键吉,*
(1河南师范大学化学化工学院,河南省绿色化学重点实验室,河南 新乡 453007;2河南师范大学,精细化学品绿色制造河南省协同创新中心,绿色化学介质与反应教育部重点实验室,河南 新乡 453007)
离子液体表面活性剂在水溶液中的自组装及其调控研究进展
王慧勇1,2李虹培1,2崔国凯1,2李志勇1,2王键吉1,2,*
(1河南师范大学化学化工学院,河南省绿色化学重点实验室,河南 新乡 453007;2河南师范大学,精细化学品绿色制造河南省协同创新中心,绿色化学介质与反应教育部重点实验室,河南 新乡 453007)
离子液体表面活性剂在化学合成、材料制备和环境污染控制等方面的应用与它们在水溶液中的自组装及其微观结构密切相关。因此,研究离子液体表面活性剂在水溶液中的自组装行为具有重要的意义。本文重点综述了阳离子的结构、阴离子的类型、外加电解质、有机添加剂、环境因素(温度、溶液pH值和光)等对离子液体表面活性剂在水中的自组装行为以及对组装体结构影响的研究进展,总结了这些因素对离子液体表面活性剂在水中自组装的调控规律,展望了该领域的发展方向及面临的挑战。
离子液体表面活性剂;水溶液;自组装;胶束;囊泡;调控
1 引 言
离子液体是由有机阳离子和无机或有机阴离子组成的一类液体盐,它的熔点一般低于100 °C。与传统的挥发性有机溶剂相比,离子液体具有独特的物理化学性质:几乎无蒸汽压、不可燃、热分解温度高、液态温度范围大(–200 – 300 °C),电化学窗口宽,对无机、有机和聚合物材料均有很好的溶解能力,结构和性质可调等1,2。正是这些优良的物理化学性质使离子液体在化学合成3、生物催化转化4,5、电化学6,7、分析分离科学8,9、生物质溶解和材料制备10–12等领域得到了广泛的应用。在这些应用过程中,离子液体表现出了显著的离子液体效应,这与离子液体的阴、阳离子的特性、离子液体与其他组分之间的相互作用以及这些体系中离子液体的微观结构密切相关13–15。在许多情况下,离子液体的功能正是通过它们在体系中所特有的微观结构来实现的。近年来的研究发现,许多离子液体具有两亲性,当烷基链较长时(称为离子液体表面活性剂)能够在水溶液中发生自组装,并且离子液体簇集体的形成在材料制备、催化合成、环境污染控制等方面发挥着重要的作用16,17。因此,研究离子液体表面活性剂在水中的自组装行为及其簇集体结构的调控对于深入认识离子液体效应具有重要的科学意义,并引起了人们越来越多的关注18–20。本文综述了阳离子的结构、阴离子的类型、外加电解质、有机添加剂、环境因素(温度、溶液pH值和光)等对离子液体表面活性剂在水中自组装行为以及簇集体结构调控的研究进展,阐述了离子液体表面活性剂簇集体结构的形成机理,总结了各种因素对离子液体表面活性剂簇集体结构的调控规律,并展望了该领域的发展方向和面临的挑战。
2 离子液体表面活性剂在水中自组装行为的调控
为了深入理解离子液体表面活性剂在水中自组装的主要驱动力,利用簇集体形成的标准吉布斯自由能、焓和熵等热力学参数对它们在水中的自组装过程进行热力学分析是非常必要的21。根据胶束化质量作用模型或相分离模型,簇集体形成的热力学参数可以通过离子液体表面活性剂的临界簇集浓度(CAC)及其随温度的变化关系推导出来22,23。离子液体表面活性剂的标准簇集自由能可以用下式计算:
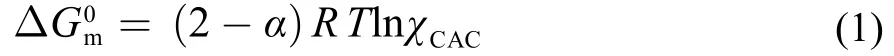
这里χCAC是用摩尔分数表示的临界簇集浓度,α是簇集体的离子化度。簇集体形成的标准焓可以由Gibbs-Helmholtz方程获得:
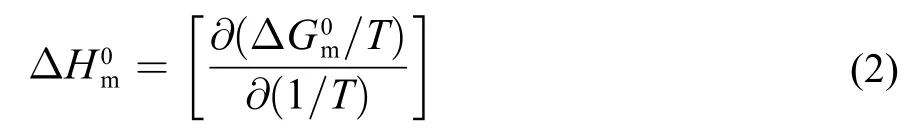
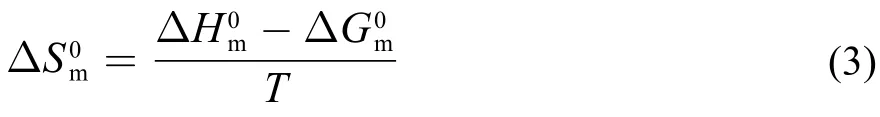
2.1 阳离子的结构对离子液体表面活性剂在水中自组装行为的调控
与常见的两亲分子类似,离子液体表面活性剂的亲/疏水性可以通过阳离子烷基链的长度、阳离子头基的类型和阴离子的特性进行调控。亲/疏水性的变化将会引起离子液体表面活性剂在水中自组装特征参数(如临界簇集浓度CAC、平均聚集数Nagg和标准簇集自由能等)的变化。因此,离子液体表面活性剂的阳离子的结构对于离子液体在水中自组装的调控引起了人们的高度关注。
对于直链表面活性剂同系物,它们的CAC值遵循Stauf-Klevens经验公式24,即lgCAC与烷基链上的碳原子数呈线性关系:

式中,nc为疏水烷基链上的碳原子数;A为常数,其值与疏水链的性质有关;B也是常数,表示一个-CH2基团对CAC值的贡献。表1列出了一些典型的传统表面活性剂和离子液体表面活性剂同系物的A和B值20,24–29。可以看出,对于非离子型的两亲分子(烷基聚氧乙烯醚、烷基二甲基氧化铵)、单离子头基的石油磺酸盐和阴离子为二价离子的烷基盐(如烷基硫酸盐),其B值分别为0.5、0.28–0.3和0.2530。对于A值而言,不同的表面活性剂相差较大;但B值接近lg2,这表明表面活性剂分子每增加一个-CH2基团,它的CAC值降为原来的1/2。据此,我们可以估算不同烷基链长度的表面活性剂的CAC值31。对于离子液体表面活性剂,其CAC值随烷基链碳原子数的关系也可以用方程(4)来描述。从表1可以看出,它们的B值与季铵盐氯化物和季铵盐溴化物的数值很接近。因此,我们可以利用传统表面活性剂的B值来预测离子液体表面活性剂同系物的CAC值。
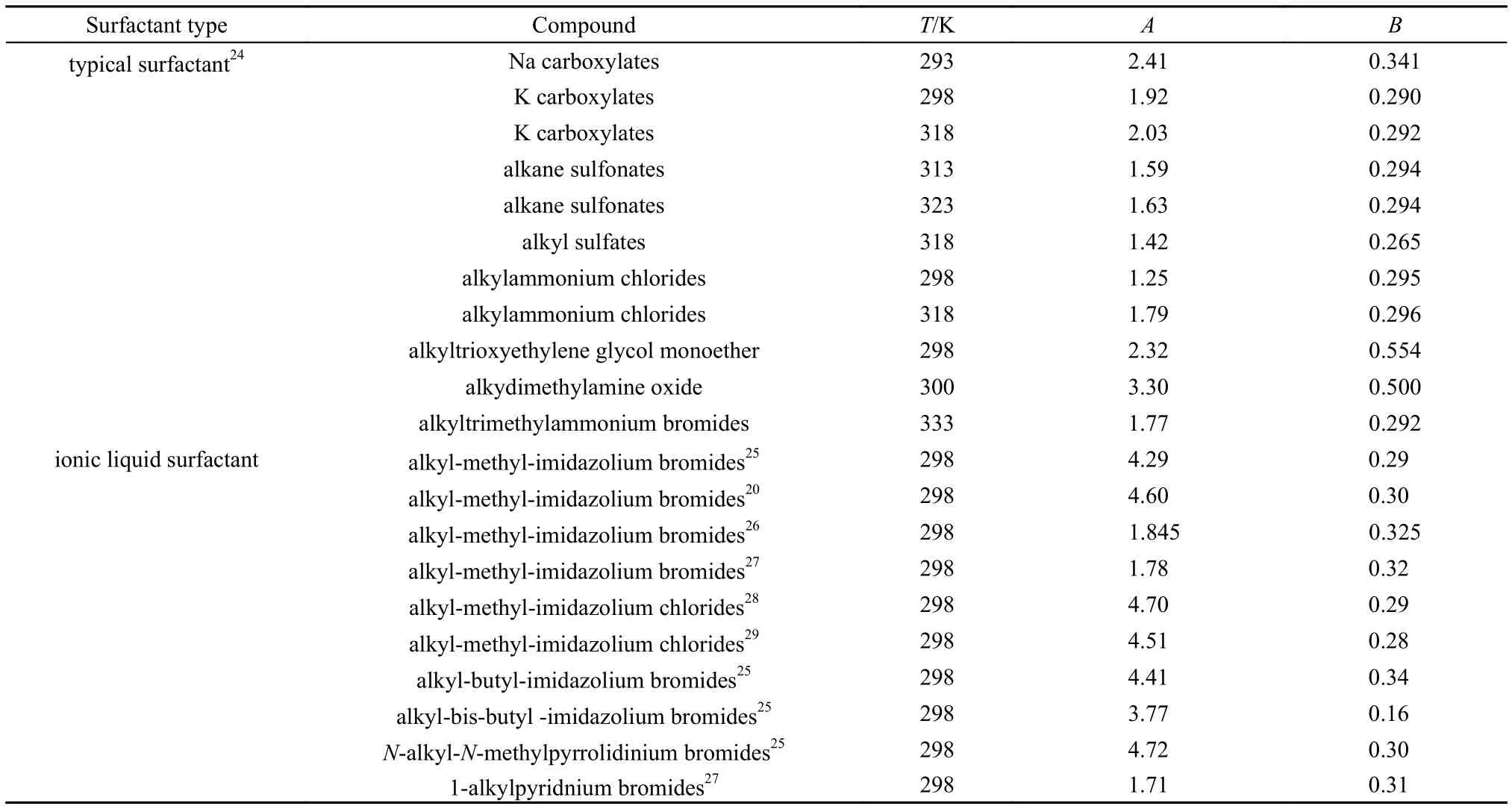
表1 一些典型的传统表面活性剂和离子液体表面活性剂同系物的A和B值Table 1 Values of A and B for various traditional surfactants and ionic liquid surfactants
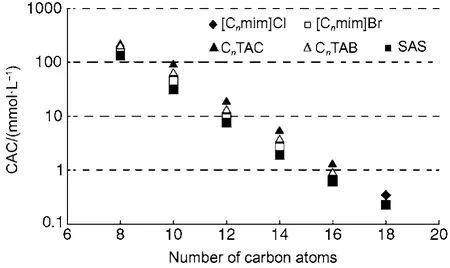
图1 298.15 K时一些离子液体表面活性剂和传统阳离子表面活性剂的临界簇集浓度(CAC)值与烷基链上碳原子数的关系32Fig.1 Critical aggregation concentration (CAC)dependence on the number of carbon atoms in alkyl chains of some ionic liquid surfactants and traditional cationic surfactants at 25 °C32
为了更好地了解离子液体表面活性剂的CAC值与一些代表性传统阳离子表面活性剂的差别,图1列出了咪唑氯化物([Cnmim]Cl),咪唑溴化物([Cnmim]Br),烷基三甲基氯化铵(CnTAC),烷基三甲基溴化铵(CnTAB)和烷基磺酸钠(SAS)在水溶液中的CAC值。可以看出,当烷基链上的碳原子数相同时,[Cnmim]Br的CAC值介于CnTAB和SAS的CAC值之间。
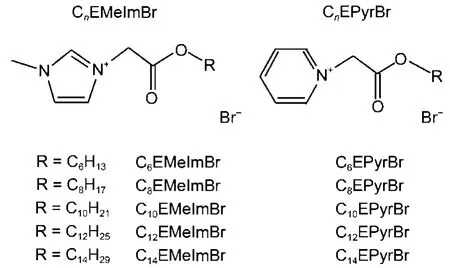
图2 酯基功能化的咪唑离子液体表面活性剂(CnEMeImBr)和吡啶离子液体表面活性剂(CnEPyrBr)的结构式Fig.2 Structures of the ester-functionalized imidazolium(CnEMeImBr) and pyridinium (CnEPyrBr) ionic liquid surfactants
离子液体阳离子烷基链上取代基团的引入将会对离子液体表面活性剂在水中的自组装有一定的影响。Garcia等33研究了在烷基链末端酯基功能化的咪唑类离子液体表面活性剂 (CnEMeImBr)和吡啶类离子液体表面活性剂 (CnEPyrBr)(结构式见图2)在水中的自组装行为。他们发现这些离子液体表面活性剂的CAC值大约是相应无酯基官能团离子液体表面活性的1/2–1/3。这可能是由于酯基的引入,降低了头基之间的静电排斥作用,有利于离子液体表面活性剂自组装的形成(CAC值降低)。Wang等34研究了烷基链末端羧基取代的N-烷基-N′-羧基咪唑溴化物[N-Cn-N′-CO2H-Im]Br (n = 10, 12, 14)在水中的自组装行为,表明这些离子液体表面活性剂的CAC值比碳原子数相同但无羧基官能团的相应的离子液体表面活性剂低。
阳离子头基的类型对离子液体表面活性剂的自组装也有着重要而复杂的影响。因为阳离子本身具有两种相反的作用35:(i)头基间的静电排斥作用、水化、空间位阻;(ii)由于降低水与碳氢链的接触而产生的烃-烃吸引相互作用。阳离子的疏水性愈强,愈有利于离子液体表面活性剂在水中的自组装;阴、阳离子间较强的相互作用能够降低头基间的排斥作用和离子液体表面活性剂的CAC值。相反,如果阳离子的空间位阻较大,将不利于离子液体表面活性剂的自组装,从而升高它的CAC值。为了检验这些因素对自组装行为的影响,我们课题组36系统地研究了具有不同阳离子结构的离子液体表面活性剂4-甲基-1-辛基吡啶溴化物(4m-[C8py]Br)、1-甲基-1-辛基四氢吡咯溴化物([C8mpyr]Br)和1-正辛基-3-甲基咪唑溴化物([C8mim]Br)在水中的自组装行为。实验结果表明,这些离子液体表面活性剂的CAC值依如下顺序增大:4m-[C8py]Br < [C8mim]Br < [C8mpyr]Br。该顺序是由阳离子的疏水性、阴阳离子之间的相互作用和阳离子的空间位阻排斥效应共同决定的。
据Blesic等37报道,3-甲基-1-十二烷基吡啶溴化物([C12mpy]Br)和1-甲基-1-十二烷基哌嗪溴化物([C12mpip]Br)在水中的CAC值几乎相同。这可能是[C12mpip]Br的两种相互补偿效应所造成的。一方面,与电荷离域的[C12mpy]+阳离子相比,[C12mpip]+能与Br–发生较强的静电相互作用,从而使得它在水中具有较低的CAC值。另一方面,由于这两种阳离子具有不同的几何构型和体积,其中[C12mpip]+比具有船式或椅式结构的[C12mpy]+有更大的几何空间,从而使[C12mpip]+阳离子具有较高的CAC值。
簇集体的离子化度(α)是表征反离子与簇集体缔合能力的一个参数。通常,在很稀的水溶液(浓度小于临界簇集浓度)中两亲分子是完全解离的,但是当簇集体形成后,部分反离子与簇集体发生缔合,从而影响两性分子的自组装。α值可以由电导–浓度曲线中高于CAC的直线斜率和低于CAC的直线斜率之比得到38。较小的α值表示反离子与簇集体有较强的缔合。对于离子液体表面活性剂,其α值通常随着烷基链长度的增加而稍微减小。
对于Gemini离子液体表面活性剂,它们的CAC值与连接两个阳离子的烷基链的长度和阳离子上烷基链的长度有关。一般情况下,连接两个阳离子的烷基链的增长导致CAC值线性降低。同样,阳离子上烷基链的增长也对CAC值产生相似的效应25,40–43。Zhang等44研究了Gemini离子液体表面活性剂1,1′-(丁烷-1,4-二取代)双(1-烷基吡啶)溴化物[Cnpy-4-Cnpy][Br2] (n = 10, 12, 14)在水中的自组装行为,表明与含有相同烷基链长度的吡啶离子液体[Cnmpy]Br相比,该Gemini离子液体表面活性剂具有较高的表面活性,较低的CAC值和较高的α值。Gemini离子液体表面活性剂在水中的自组装是焓控制过程,而[Cnmpy]Br在水中的自组装在低温时是熵控制过程,高温时是焓控制过程。
Jiao45和Blesic46等研究了Catanionic离子液体表面活性剂1-烷基-3-甲基咪唑烷基硫酸盐[Cnmim][CmSO4] (n = 6, 8, 10; m = 12 和n = 4; m = 10, 14)在水中的自组装行为。实验结果表明,这些离子液体表面活性剂比相应的阴离子或阳离子类似物有较高的表面活性和较低的CAC值,阳离子或阴离子烷基链的增长都会降低这些离子液体表面活性剂的CAC值,并且CAC的对数值与阴、阳离子上烷基链的碳原子数的总和存在很好的线性关系。
2.2 阴离子的类型对离子液体表面活性剂在水中自组装行为的调控
离子液体阴离子的特性决定了离子液体的水溶性和阴、阳离子之间的相互作用。因此,阴离子的特性将影响离子液体表面活性剂在水中的自组装行为。我们课题组较系统地研究了离子液体表面活性剂的阴离子对自组装行为的影响36。研究结果表明,对于阳离子为[C8mim]+的离子液体表面活性剂,其CAC值和α值均按照以下顺序增加:[CF3COO]–< [NO3]–< Br–< Cl–< [CH3COO]–。此顺序与阴离子对传统阳离子表面活性剂簇集行为影响的Hofmeister顺序相一致47,48。阴离子在Hofmeister顺序中的位置与阴离子的水化半径/极化率、疏水性和体积的大小有着密切的关系。具有较大水化半径的阴离子通常具有较强的极化能力,能够有效地增强阴离子在簇集体表面的键合,降低头基之间的静电排斥,从而增强离子液体自组装的趋势,降低CAC值和a值。但是,[C8mim][CF3SO3]和[C8mim][ClO4]在水中没有发生自组装。因为在这些离子液体表面活性剂发生自组装之前,水溶液已经分为两相。这说明这两种阴离子都有较弱的水化能力,使离子液体在水中有较低的溶解度。Blesic等28也观察到,当 [C10mim]Cl中的Cl–阴离子被[NTf2]–或者[PF6]–取代后,这些离子液体表面活性剂不能在水中形成胶束,因为它们在水中的溶解度较低,自组装之前已经发生相分离。
离子液体的阴离子对表面活性剂在水中的标准簇集自由能的影响遵循Hofmeister顺序。图3描述了离子液体表面活性剂的标准簇集自由能与其阴离子的水化自由能之间的关系49。表明阴离子的疏水性愈强,离子液体表面活性剂愈容易发生自组装。这是由于阴离子较强的疏水性增强了它与阳离子簇集体的键合作用,降低了阳离子头基之间的排斥作用。Bunton和Cowell50在研究十六烷基三烷基溴化铵和甲基萘-2-磺酸盐的相互作用时,也得到了类似的结果。阴离子对聚集数(Nagg)的影响也与阴离子的亲/疏水性有着密切的影响。阴离子的疏水性愈强,离子液体的平均簇集数越大,这与传统表面活性剂胶束增长的反离子效应相类似51。

图3 离子液体表面活性剂的标准簇集自由能与其阴离子水化自由能的线性关系Fig.3 Linear plot of the standard Gibbs energy of aggregation of the ionic liquid surfactantsvs the Gibbs energy of hydration of their anions
2.3 外加电解质对离子液体表面活性剂在水中自组装行为的调控
据文献报道,水中电解质的存在会显著影响水的结构和离子型表面活性剂的水化能力,从而对离子型表面活性剂在水中的自组装产生盐析和盐溶效应。当离子型表面活性剂的单体被电解质盐析后,它的CAC值降低;反之,它的CAC值增加48,52。由于离子液体表面活性剂在某些方面与离子型表面活性剂相类似,它们在水中的自组装行为也可以通过外加电解质的方式来调控。
一般情况下,无机盐对离子液体表面活性剂在水中自组装行为的影响与无机盐和离子液体表面活性剂的阴、阳离子的特性有着密切的关系。人们研究发现28,53–57,大部分无机盐的加入降低了离子液体表面活性剂在水中的CAC值。随着无机盐浓度的增加,CAC值进一步降低。这是由于这些无机盐通过与胶束的双电层相互作用,屏蔽了胶束表面的电荷,降低了离子液体头基间的排斥作用,促进了离子液体表面活性剂的自组装。这一规律与无机盐对十二烷基三甲基溴化铵和十六烷基三甲基溴化铵58的影响规律类似。
我们课题组59利用电导、荧光和动态光散射技术较系统地研究了15种无机盐对 [C10mim]Br在水中自组装行为的影响。实验结果也表明,无机盐的加入较显著地降低了[C10mim]Br在水中的CAC值。无机盐的阴离子对离子液体表面活性剂CAC值的影响按照以下顺序递增:这一顺序与无机盐阴离子的Hofmeister顺序基本一致,为我们提供了无机盐对离子液体表面活性剂自组装行为影响的定性描述方法60,61。对于给定的无机盐,离子液体表面活性剂CAC值的对数随着无机盐浓度的增加而线性减小,可以用传统表面活性剂的盐效应方程来描述:
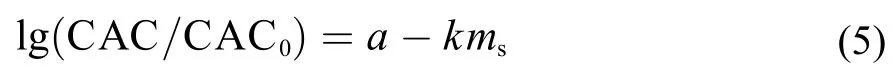
式中,CAC和CAC0分别表示有盐和无盐存在时离子液体表面活性剂的临界簇集浓度,s表示所添加盐的浓度,a和k是方程的待定参数,其中k称之为盐效应常数,可通过现代盐效应理论来预测。
另外,无机盐对离子液体的阴离子在簇集体表面的键合度、簇集自由能和簇集数的影响也基本上遵循Hofmeister顺序60。对于给定的无机盐,阴离子的键合度和簇集数均随无机盐浓度的增加而增大,簇集体的尺寸随着无机盐中阴离子疏水性的增强而增大。这是因为无机盐中疏水性较强的阴离子在簇集体表面具有较强的键合作用,降低了头基之间的排斥作用,促进了离子液体表面活性剂的生长。
然而,FeBr3和AlBr3对离子液体表面活性剂的簇集具有盐溶效应60。我们知道,Br–和FeBr3(或者AlBr3)在水中能形成具有四面体结构的配合物与Br–相比,[FeBr4]–和[AlBr4]–具有较大的半径和较小的电荷密度,它们不能有效降低阳离子头基之间的排斥作用,但能与阳离子的疏水部分发生作用,从而降低了离子液体表面活性剂的簇集能力,增大了它们的CAC值。
此外,Sadeghi和Golabiazar62研究了有机电解质(Me4NBr、Et4NBr、Pr4NBr、Bu4NBr、Me4NCl、Me4NI)对[C12mim]Br在水中自组装行为的影响。他们观察到,这些有机电解质也能降低[C12mim]Br在水中的CAC值,对[C12mim]Br的自组装具有盐析效应。但是,有机阳离子的盐析效应较弱,而无机阴离子对离子液体表面活性剂的自组装行为具有较大的影响。阴离子对离子液体表面活性剂自组装行为的促进能力以I–> Br–> Cl–顺序增强,而[Me4N]+、[Et4N]+、[Pr4N]+和[Bu4N]+具有相似的盐析效应强度。这再次表明,疏水性较强的阴离子有利于促进离子液体表面活性剂在水中的自组装。
Gu等63研究了短链的离子液体1-丁基-3-甲基咪唑甲基磺酸盐 ([C4mim][Msa])、1-丁基-3-甲基咪唑苯磺酸盐([C4mim][Bsa])和1-丁基-3-甲基咪唑萘磺酸盐([C4mim][Nsa])对[C12mim]Br在水中自组装行为的影响。他们的研究结果表明,这三种短链的离子液体在水溶液中的作用与普通的电解质类似,都能够促进[C12mim]Br在水中的自组装,并且以[C4mim][Nsa] > [C4mim][Bsa] > [C4mim][Msa]顺序增强。这里,[Msa]–、[Bsa]–和[Nsa]–阴离子的疏水性起到了非常重要的作用。
2.4 有机添加剂对离子液体表面活性剂在水中自组装行为的调控
胶束液相色谱(MLC)是以胶束水溶液为流动相的一种色谱技术64–67。当有机溶剂加入后,往往形成三相体系65,67,68。然而,目前只有少数表面活性剂体系被成功地应用到MLC技术中66,69。因此,有机溶剂对离子液体表面活性剂在水中自组装行为影响的研究不仅对胶体界面科学有着重要的意义,而且有可能扩展离子液体在胶束液相色谱中的应用。
Pino等70用表面张力研究了[C16C4im]Br和[C12C12im]Br在(水 + 有机溶剂)体系中的胶束化性质。所用有机溶剂包括甲醇、正丙醇、正丁醇、正戊醇和乙腈。研究结果表明,随着有机溶剂的加入,增加了这两种离子液体表面活性剂的CAC值和单个表面活性分子的最小表面积(Amin),降低了最大表面过剩浓度(Γmax)、吸附效率(pC20)和表面张力降低效率(πcmc)。有机溶剂对[C12C12im]Br的影响强于[C16C4im]Br。
我们课题组71较系统地研究了有机添加剂(乙二醇、二甲亚砜、二甘醇、三甘醇、甲酰胺、乙腈、甲醇、乙醇、正丙醇、丙酮)对[C12mim]Br在水中自组装行为的影响。表明随着有机添加剂在水中含量的增加,[C12mim]Br在水中的CAC、α和值均增加,而聚集数和簇集体的尺寸均减小。这些结果可以通过离子液体表面活性剂的烷基链与水-有机物混合溶剂的疏溶剂相互作用来解释:水-有机物混合溶剂能够更好地溶剂化离子液体的烷基链,使表面活性剂的自组装能力下降,直接导致CAC、α和值增加和聚集数、聚集体尺寸减小。有机溶剂对 [C12mim]Br在水溶液中CAC的影响规律与有机溶剂对十二烷基三甲基溴化铵72和十四烷基三甲基溴化铵等传统表面活性剂的影响规律类似73。另外,离子液体表面活性剂CAC的对数值与水-有机物混合溶剂的疏溶剂参数(Sp)之间存在线性关系,可以用下列方程来描述71:
2.5 体系温度对离子液体表面活性剂在水中自组装行为的调控
温度对离子液体表面活性剂在水溶液中CAC的影响主要体现在两个方面:一方面,温度升高减弱了阳离子亲水头基的水合作用,可以促进胶束的形成,使CAC值减小; 另一方面,温度升高破坏了疏水烷基链周围的水合结构,不利于胶束的形成,使CAC值增大。研究工作者利用电导方法测定了不同温度下咪唑类离子液体表面活性剂[Cnmim]X (n = 12–16)在水中的CAC值22,发现该离子液体表面活性剂的CAC值随温度的变化呈“U型”,其CAC的最低值所对应的温度(Tmin)接近室温74–77。他们认为,Tmin值与烷基链的疏水性和反离子的性质密切相关,疏水性较强的两亲分子具有更低的Tmin值。图4描述了咪唑离子液体表面活性剂簇集体形成的热力学参数随温度的变化关系。可以看到,首先随着温度的升高而降低,然后在20–25 °C附近由正值变为负值。这意味着,这些离子液体表面活性剂簇集体的形成过程在低温时是吸热过程,在高温时是放热过程。其次,随着温度的升高而增加,在25–30 °C附近与相交。这表明当体系温度低于交点温度时,熵变对的负值起着决定性作用,而焓变在温度高于该温度时对的负值起主导作用。即咪唑离子液体表面活性剂簇集体的形成在低温下是熵控制过程,而在高温下是焓控制过程。这与吡啶类溴化物[Cnpy]Br (n = 12, 14, 16)在水中簇集体形成的热力学控制特性相同78。
3 离子液体表面活性剂在水中簇集体的结构
离子液体在化学反应和材料制备中表现出的独特性质与离子液体在溶液中的微观结构密切相关。因此,离子液体表面活性剂在溶液中的簇集体的微观结构研究引起人们的高度重视,并且利用中子散射(SANS)、透射电镜(TEM)、核磁共振(NMR)和分子动力学模拟(MD)等手段进行了卓有成效的研究。与传统的表面活性剂类似,常见的离子液体表面活性剂簇集体的结构包括胶束和囊泡等。
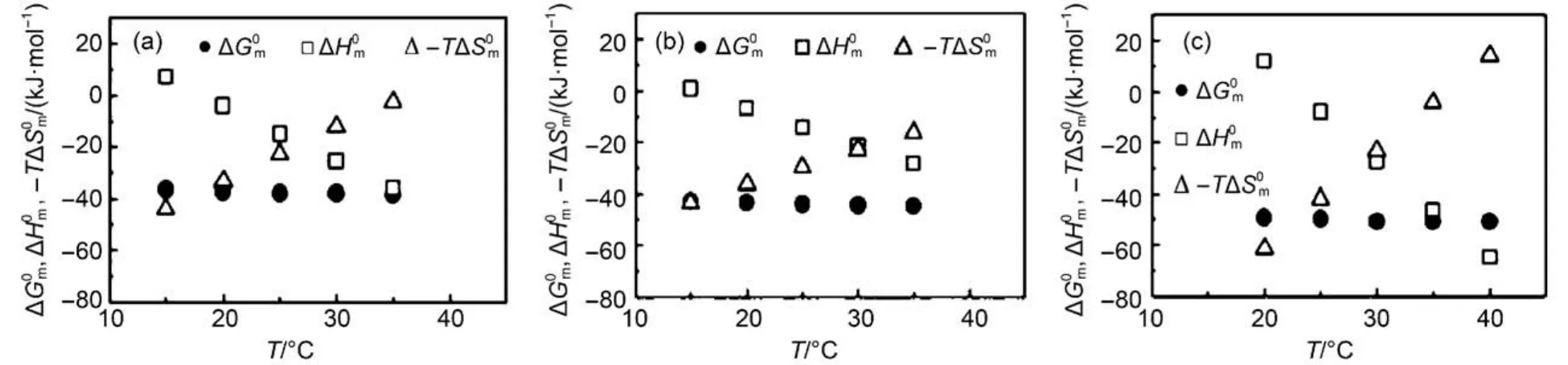
图4 咪唑离子液体表面活性剂簇集体形成过程的热力学参数随温度的变化关系22Fig.4 Variation of the thermodynamic parameters for the micelle formation of some imidazolium-based ionic liquid surfactants with temperature22
3.1 胶 束
胶束是分子有序组合体最基本和最常见的形式。近年来,关于离子液体表面活性剂在水溶液中自组装形成胶束的研究报道较多。Bower等18和Goodchild等79利用SANS技术研究了[Cnmim]X (n = 2, 4, 6, 8, 10; X = Cl, Br)、[C8mim]I和[C4mim][BF4]在水中聚集体的微观结构,发现[C2mim]Br和[C4mim]Br不能形成胶束;当离子液体的浓度高于CAC时,[C4mim][BF4]形成了多分散性的球形簇集体,[C8mim]X (X = Cl, Br, I)或[C10mim]Br以较小的球形簇集体存在;随着浓度的进一步增加,[C10mim]Br的胶束变得更扁长。这些胶束结构已被NMR80和MD81,82研究所支持:在这些簇集体中,烷基链被埋在簇集体的内部,避免与水的接触;而极性的咪唑基团则位于簇集体的表面,并暴露于水中。我们课题组利用TEM和动态光散射技术研究了[C8mim]X (X = Cl, Br, NO3, CH3COO, CF3COO)、[C8mpyr]Br和4m-[C8py]Br在水中簇集体的结构36。实验结果表明,这些离子液体表面活性剂都能形成球形胶束,阴、阳离子的结构对胶束的形态影响很小,但是簇集体的尺寸随着离子液体阴离子疏水性的增加而增大。总之,离子液体表面活性剂疏水烷基链的长度、阴离子的特性、阳离子头基的结构、溶液的浓度及电荷密度等都会影响胶束的形成。
3.2 囊 泡
囊泡是某些两亲分子分散在水中自发形成的一类具有封闭双层结构的聚集体。囊泡不但可以用来模拟生物细胞膜和多种生物生理过程、药物的封装及输送等,而且还能够提供化学反应所需要的“微环境”83。人们发现,离子液体表面活性剂与一些传统的表面活性剂在水中复配能够形成囊泡。例如,Singh 等84报道了由烷基吡啶类离子液体表面活性剂[C6Py]Br或[C6Py][BF4]与十二烷基硫酸钠(SDS) 在水溶液中复配形成的囊泡;与[C6Py][BF4]/SDS 体系相比,在[C6Py]Br/SDS 体系中形成的囊泡具有较大的尺寸;随着体系中阴离子表面活性剂SDS浓度的增加,复配体系中的囊泡数量也随之增多。Yuan等85观察到,在[C12mim]Br/ SDS和[C12mPyrr]Br/十二烷基硫酸铜四水化合物( Cu(DS)2·4H2O)复配体系中也可以形成囊泡。
离子液体表面活性剂也可以与另外一种离子液体在水中复配形成囊泡。Rao等86的实验结果表明,两种离子液体表面活性剂[C4mim][C8OSO3]和[C8mim]Cl在水中以一定的比例混合能够形成胶束和囊泡。此外,当浓度达到一定值后Catanionic型离子液体表面活性剂在水中也可以形成囊泡。例如,据Rao等87报道,由1,3-二烷基咪唑阳离子和烷基磺酸阴离子构成的离子液体[CnH2n+1mim][CmH2m+1OSO3](n = 4, 6, 8;m = 8, 12)在水中能够发生簇集,当n = 4, 6及m = 8时, 离子液体表面活性剂形成了胶束;当n = 8及m = 8, 12时,则形成了囊泡。Villa等88也报道了Catanionic型离子液体表面活性剂(由苄基-n-己基癸基二甲基季铵阳离子和二(2-乙基)己基磺化琥珀酸阴离子构成)在水中单壁囊泡的形成。最近的研究工作表明,含有双尾烷基链的离子液体表面活性剂在水中也能形成囊泡。例如,Rao等89制备了二(2-乙基)己基磺化琥珀酸盐离子液体表面活性剂(阳离子为[C4mim]+、脯氨酸异丙酯阳离子[ProC3]+、胆碱阳离子[Cho]+、胍阳离子[Gua]+)。这些离子液体表面活性剂在水中都能形成囊泡,并且形成囊泡的浓度按照 [ProC3]+<[C4mim]+< [Gua]+< [Cho]+顺序增大。该顺序与阳离子的特性有关。

图5 水溶液中[C12mim]Br在不同浓度下聚集体的TEM照片91Fig.5 TEM images for the aggregates of [C12mim]Br in aqueous solution at different concentrations91
但是,对于传统的单尾、离子型的小分子表面活性剂,在没有任何添加剂(如无机盐、表面活性剂)存在时,它们自身不能在水溶液中形成囊泡90。那么单尾离子液体表面活性剂在没有任何添加剂存在下能否形成囊泡?带着这个问题,我们以[Cnmim]Br (n = 6, 8, 10, 12, 14)为研究对象,采用表面张力、1H NMR、动态光散射、透射电镜和小角X射线光散射等技术研究了离子液体表面活性剂在水溶液中的自组装结构91,并首次观察到在无任何添加剂存在的情况下,[Cnmim]Br (n = 10, 12, 14)在水中形成了单层囊泡,这是由离子液体表面活性剂独特的性质——咪唑阳离子较小的电荷密度和头基间的π–π相互作用所决定的。随着浓度的增大,离子液体表面活性剂在水中的自组装结构经历着从球形胶束→柱状胶束→单层囊泡的转变,而且该转变过程是可逆过程(图5)。这类离子液体表面活性剂扩展了囊泡形成的体系。随后,Shi等92也表明,在无任何添加剂存在的情况下,单尾离子液体表面活性剂1-十二烷基-3-甲基咪唑-β-萘酚磺酸盐[C12mim][Nsa]在水中也形成了囊泡,并且随着离子液体浓度的增加,簇集体的结构经历着从胶束→单、多壁囊泡→双层结构→多层液晶相的转变(如图6所示)。Du等93也报道在无任何添加剂存在的情况下,单尾的离子液体表面活性剂十二烷基三甲基溴化铵(DTAB)在毛玻璃调节下能够在水中自发地形成囊泡。

图6 [C12mim][Nsa]-H2O体系相变的可能机制92Fig.6 Possible mechanism for phase change of[C12mim][Nsa]-H2O system92
3.3 离子液体表面活性剂在水溶液中胶束-囊泡转变的调控
离子液体表面活性剂在水中的浓度直接影响着它在水中组装体的结构。Rodríguez-Dafonte等94报道称,双链的离子液体表面活性剂1,3-二癸基-2甲基咪唑氯化物在水溶液中有2个CAC值,并且随着浓度的增大,离子液体表面活性剂在水中的簇集体结构首先从球形胶束转变为柱状胶束,然后再转变为层状结构。
外加电解质也可以诱导离子液体表面活性剂在水中聚集体结构的转变。据Gu等63报道,当离子液体1-丁基-3-甲基咪唑萘磺酸盐([C4mim][Nsa])被加入到[C12mim]Br的水溶液时,能够诱导[C12mim]Br的簇集体结构从胶束转变为囊泡。这是由于[C10H7SO3]–阴离子能够穿插进入[C12mim]Br簇集体的内部,降低了阳离子头基之间的静电排斥,有利于[C12mim]Br更有效地堆积。其中[C10H7SO3]–阴离子与咪唑环之间的π–π相互作用起着重要的作用。Rao等95研究表明,在水溶液中加入NaBr也能够诱导基于十二烷基苯磺酸根阴离子的离子液体表面活性剂(阳离子为1-丁基-3甲基咪唑阳离子、N-丁基吡啶阳离子和正丁基三甲基季铵阳离子)的簇集体从胶束到囊泡的转变。主要原因是NaBr的加入增强了阳离子头基与反离子之间的静电相互作用、阳离子–π和π–π相互作用,有利于簇集体的生长,从而造成簇集体的结构发生转变。
溶液pH值的改变是用来调控表面活性剂簇集体结构的一种重要手段。我们课题组设计、合成了pH响应离子液体表面活性剂[Cnmim]X (n = 10, 12, 14;X = [C6H4COOCOOK], [C6H3OHCOOSO3Na],[C6H4COOSO3Na]),并利用表面张力、1H NMR、动态光散射和透射电镜等手段研究了溶液pH值对pH响应离子液体表面活性剂在水中自组装体结构的影响96,97。实验结果表明,随着溶液pH值的改变,离子液体表面活性剂在水中的自组装体结构发生了由球形胶束→囊泡→球形胶束的可逆转变。该转变归结于由溶液pH值引起的离子液体表面活性剂阴离子的结构和亲/疏水性的改变。
与pH值、加热等外在手段相比,光具有信号易得、稳定可靠、不污染体系等优点。同时,它的光斑具有几微米的大小,可以精确地指向目标,因此常被用来调控表面活性剂在水中簇集体的结构。我们课题组合成了一类新型肉桂酸类光响应离子液体表面活性剂98。此类离子液体由咪唑类阳离子[Cnmim]+(n = 10, 12, 14, 16)和反式邻甲氧基肉桂酸[OMCA]–阴离子组成。经紫外光照射后,离子液体表面活性剂的蠕虫状胶束转变为棒状胶束。这是由于光照引起肉桂酸阴离子由反式异构体转变为顺式异构体,引起阴离子的空间位阻增强,疏水性变弱,不利于胶束的生长。随后,Bi等99设计了由[C16mim]Br/偶氮苯甲酸钠Na[AzoCOO]组成的光响应体系。该体系能够在水中形成蠕虫状胶束,光照后蠕虫状胶束变得更长、更纠缠,进而引起溶液的粘度明显地增大。主要原因是,光照后偶氮苯由反式异构体变为顺式结构,有利于苯环与咪唑环之间更有效地重叠,降低咪唑阳离子之间的静电排斥,促进胶束的生长。进一步的研究表明,[C16mim]Br和Na[AzoCOO]之间存在疏水、静电和阳离子–π等多种相互作用,其中阳离子–π相互作用是决定光照后蠕虫状胶束增长的主要驱动力。
4 结论与展望
综上分析可以看出,离子液体表面活性剂的烷基链的长度、阳离子的结构、阴离子的类型、外加电解质、有机添加剂、体系的温度、溶液的pH值和光照等对离子液体表面活性剂在水中的临界簇集浓度、离子化度、簇集体的标准吉布斯自由能、簇集数、簇集体的结构和尺寸有着重要的影响。因此,我们可以通过改变上述因素来调控离子液体表面活性剂在水中的自组装行为。但是,尽管离子液体表面活性剂在水中自组装行为的研究已取得了重要的进展,目前仍存在很多问题需要进一步研究。例如,与传统的表面活性剂相比,离子液体表面活性剂在自组装方面到底具有什么优势?离子液体表面活性剂在水中还存在那些特殊的自组装体结构?离子液体表面活性剂在水中能够形成什么样的溶致液晶?这些都是需要进一步研究的问题。因此,未来该领域的研究重点应该主要包括以下几个方面:深入研究离子液体表面活性剂的结构与簇集体结构之间的关系,探索离子液体有序组装体形成的机理及组装规律,实现组装体结构的调控、预测及新结构的形成;系统研究离子液体表面活性剂在水中形成的溶致液晶相;离子液体有序组装体的应用研究,特别是在轻工、医药、药物输送等领域中的应用。
(1)Welton, T. Chem. Rev. 1996, 99, 2071.
(2)Seddon, K. R. J. Chem. Tech. Biotechnol. 1997, 68, 351.
(3)Wasserschein, P.; Welton, T. Ionic Liquids in Syntheses; VCHWiley: Weinhein, 2003
(4)Rantwijk, F.; Lau, R. M.; Sheldon, R. A. Trends Biotechnol. 2003, 21, 131. doi: 10.1016/S0167-7799(03)00008-8
(5)Jain, N.; Kumar, A.; Chauhan, S.; Chauhan, S. M. S. Tetrahedron 2005, 61, 1015. doi: 10.1016/j.tet.2004.10.070
(6)Buzzeo, M. C.; Evans, R. G.; Compton, R. G. ChemPhysChem 2004, 5, 1106.
(7)Endres, F.; Abedin, S. Z. E. Phys. Chem. Chem. Phys. 2006, 8, 2101. doi: 10.1039/b600519p
(8)Liu, J.; Jonsson, J. A.; Jing, G. Trends. Anal. Chem. 2005, 24, 20. doi: 10.1016/j.trac.2004.09.005
(9)Zhao, H.; Xia, S.; Ma, P. J. Chem. Technol. Biotechnol. 2005, 80, 1089
(10)Zhang, S.; Sun, J.; Zhang, X.; Xin, J.; Miao, Q.; Wang, J. Chem. Soc. Rev. 2014, 43, 7838. doi: 10.1039/C3CS60409H
(11)Chen, S.; Zhang, S.; Liu, X.; Wang, J.; Wang, J.; Dong, K.; Sun, J.; Xu, B. Phys. Chem. Chem. Phys. 2014, 16, 5893.
(12)Hayes, R.; Warr, G. G.; Atkin, R. Chem. Rev. 2015, 115, 6357. doi: 10.1021/cr500411q
(13)Dupont, J. Accounts Chem. Res. 2011, 44, 1223. doi: 10.1021/ar2000937
(14)Neto, B. A. D.; Meurer, E. C.; Galaverna, R.; Bythell, B. J.;Dupont, J.; Cooks, R. G.; Eberlin, M. N. J. Phys. Chem. Lett. 2012, 3, 3435. doi: 10.1021/jz301608c
(15)Marcus, Y.; Hefter, G. Chem. Rev. 2006, 106, 4585. doi: 10.1021/cr040087x
(16)Visser, A.; Swaltowski, R. P.; Reichert, R. M.; Mayton, R.;Sheff, S.; Wierzbicki, A.; Davis, J. H.; Rogers, R. D. Environ. Sci. Technol. 2002, 36, 252.
(17)Huddleston, J. G.; Visser, A. E.; Reichert, M. W.; Willauer, H. D.; Broker, G. A.; Rogers, R. D. Green Chem. 2001, 3, 156. doi: 10.1039/b103275p
(18)Bowers, J. P.; Butts, C. J.; Martin, P. C.; Vergara-Gutierrez, M. Langmuir 2004, 20, 2191. doi: 10.1021/la035940m
(19)Miskolczy, Z.; Sebok-Nagy, K.; Biczok, L.; Gokturk, S. Chem. Phys. Lett. 2004, 400, 296. doi: 10.1016/j.cplett.2004.10.127
(20)Vanyur, R.; Biczok, L.; Miskolczy, Z. Colloids Surf. A: Physicochem. Eng. Asp. 2007, 299, 256. doi: 10.1016/j.colsurfa.2006.11.049
(21)Bai, G.; Lopes, A.; Bastos, M. J. Chem. Thermodyn. 2008, 40, 1509. doi: 10.1016/j.jct.2008.05.016
(22)Inoue, T.; Ebina, H.; Dong, B.; Zheng, L. J. Colloid Interface Sci. 2007, 314, 236. doi: 10.1016/j.jcis.2007.05.052
(23)Łuczak, J.; Hupka, J.; Thoeming, J.; Jungnickel, C. International Scientific Conference, Surfactants and Dispersed Systems in Theory and Practice; PALMA Press: Wrocław/ Ksiaz Castle, 2007.
(24)Klevens, H. B. J. Am. Oil Chem. Soc. 1953, 30, 74. doi: 10.1007/BF02635002
(25)Baltazar, Q. Q.; Chandawalla, J.; Sawyer, K.; Anderson, J. L. Colloids Surf. A: Physicochem. Eng. Asp. 2007, 302, 150. doi: 10.1016/j.colsurfa.2007.02.012
(26)Wang, J.; Wang, H.; Zhang, S.; Zhang, H.; Zhao, Y. J. Phys. Chem. B 2007, 111, 6181. doi: 10.1021/jp068798h
(27)Stepnowski, P.; Nichthauser, J.; Mrozik, W.; Buszewski, B. Anal. Bioanal. Chem. 2006, 385, 1483. doi: 10.1007/s00216-006-0577-0
(28)Blesic, M.; Marques, M. H.; Plechkova, N. V.; Seddon, K. R.;Rebelo, L. P. N.; Lopes, A. Green Chem. 2007, 9, 48.
(29)Jungnickel, C.; Łuczak, J.; Ranke, J.; Fernández, J. F.; Müller, A.; Thöing, J. Colloids Surf. A: Physicochem. Eng. Asp. 2008,316, 278. doi: 10.1016/j.colsurfa.2007.09.020
(30)Huibers, P. D. T.; Lobanov, V. S.; Katritzky, A. R.; Shah, D. O.;Karelson, M. J. Colloid Interface Sci. 1997, 187,113. doi: 10.1006/jcis.1996.4680
(31)Baker, G. A.; Pandey, S.; Pandey, S.; Baker, S. N. Analyst 2004,12, 890.
(32)Łuczaka, J.; Hupkaa, J; Thöing, J.; Jungnickel, C. Colloids Surf. A: Physicochem. Eng. Asp. 2008, 329, 125. doi: 10.1016/j.colsurfa.2008.07.012
(33)Garcia, M. T.; Ribosa, I.; Perez, L.; Manresa, A.; Comelles, F. Langmuir 2013, 29, 2536. doi: 10.1021/la304752e
(34)Wang, X. Q.; Yu, L.; Jiao, J. J.; Zhang, H. N.; Wang, R.; Chen, H. J. Mol. Liq. 2012, 173, 103. doi: 10.1016/j.molliq.2012.06.023
(35)Brady, J. E.; Evans, D. F.; Warr, G. G.; Grieser, F.; Niham, B. W. J. Phys. Chem. 1986, 90, 1853. doi: 10.1021/j100400a024
(36)Wang, H.; Wang, J.; Zhang, S.; Xuan, X. J. Phys. Chem. B 2008,112, 16682. doi: 10.1021/jp8069089
(37)Blesic, M.; Lopes, A.; Melo, E.; Petrovski, Z.; Plechkova, N. V.;Canongia Lopes, J. N.; Seddon, K. R.; Rebelo, L. P. N. J. Phys. Chem. B 2008, 112, 8645. doi: 10.1021/jp802179j
(38)Zana, R. Langmuir 1996, 12, 1208. doi: 10.1021/la950691q
(39)Tokuda, H.; Hayamizu, K.; Ishii, K.; Suan, M. D. A. B. H.;Watanabe, M. J. Phys. Chem. B 2005, 109, 6103. doi: 10.1021/jp044626d
(40)Ao, M.; Huang, P.; Xu, G.; Yang, X.; Wang, Y. Colloid Polym. Sci. 2009, 287, 395. doi: 10.1007/s00396-008-1976-x
(41)Bhadani, A.; Singh, S. Langmuir 2011, 27, 14033. doi: 10.1021/la202201r
(42)Kamboj, R.; Singh, S.; Bhadani, A.; Kataria, H.; Kaur, G. Langmuir 2012, 28, 11969. doi: 10.1021/la300920p
(43)Palchowdhury, S.; Bhargava, B. L. Phys. Chem. Chem. Phys. 2015, 17, 11627. doi: 10.1039/C5CP00873E
(44)Zhang, S.; Yan, H.; Zhao, M.; Zheng, L. J. Colloid Interface Sci. 2012, 372, 52. doi: 10.1016/j.jcis.2012.01.040
(45)Jiao, J.; Han, B.; Lin, M.; Cheng, N.; Yu, L.; Liu, M. J. Colloid Interface Sci. 2013, 412, 24. doi: 10.1016/j.jcis.2013.09.001
(46)Blesic, M.; Swadźba-Kwaśny, M.; Holbrey, J. D.; Lopes, J. C.;Seddonab, K. R.; Rebelo, L. P. N. Phys. Chem. Chem. Phys. 2009, 11, 4260. doi: 10.1039/b822341f
(47)Luo, G.; Qi, X.; Han, C.; Liu, C.; Gui, J. J. Surfact. Deterg. 2013,16, 531. doi: 10.1007/s11743-012-1431-3
(48)Sepúlveda, L.; Cortés, J. J. Phys. Chem. 1985, 89, 5322. doi: 10.1021/j100270a040
(49)Marcus, Y. J. Chem. Soc. Faraday Trans. 1991, 87, 2995. doi: 10.1039/ft9918702995
(50)Bunton, C. A.; Cowell, C. J. Colloid Interface Sci. 1988, 122, 154. doi: 10.1016/0021-9797(88)90298-6
(51)Abdel-Rahem, R. Adv. Colloid Interface Sci. 2008, 141, 24. doi: 10.1016/j.cis.2008.02.002
(52)Shaw, D. J. Introduction to Colloid and Surface Chemistry;Butterworth, Heinemann: Oxford, 1992.
(53)Dong, B.; Li, N.; Zheng, L.; Yu, L.; Inoue, T. Langmuir 2007,23, 4178.
(54)Dong, B.; Zhao, X.; Zheng, L.; Zhang, J.; Li, N.; Inoue, T. Colloids Surf. A: Physicochem. Eng. Asp. 2008, 317, 666. doi: 10.1016/j.colsurfa.2007.12.001
(55)Vaghela, N. M.; Sastry, N. V.; Aswal, V. K. Colloids Surf. A: Physicochem. Eng. Asp. 2011, 373, 101. doi: 10.1016/j.colsurfa.2010.10.031
(56)Ghasemian, E.; Najafi, M.; Rafati, A. A.; Felegari, Z. J. Chem. Thermodyn. 2010, 42, 962. doi: 10.1016/j.jct.2010.03.007
(57)Golabiazar, R.; Sadeghi, R. J. Chem. Thermodyn. 2014, 76, 29. doi: 10.1016/j.jct.2014.03.001
(58)Larsen, J. W.; Magidl, L. J. J. Am. Chem. Soc. 1974, 96, 5774. doi: 10.1021/ja00825a013
(59)Wang, H.; Feng, Q.; Wang, J.; Zhang, H. J. Phys. Chem. B 2010,114, 1380. doi: 10.1021/jp910903s
(60)Anacker, E. W.; Ghose, H. M. J. Am. Chem. Soc. 1968, 90, 3161. doi: 10.1021/ja01014a034
(61)Freire, M. G.; Carvalho, P. J.; Silva, A. M. S. J. Phys. Chem. B 2009, 113, 202. doi: 10.1021/jp8080035
(62)Sadeghi, R.; Golabiazar, R. J. Mol. Liq. 2014, 197, 176. doi: 10.1016/j.molliq.2014.04.034
(63)Gu, Y.; Shi, L.; Cheng, X.; Lu, F.; Zheng, L. Langmuir 2013, 29, 6213. doi: 10.1021/la400497r
(64)Armstrong, D. W.; Henry, S. J. J. Liq. Chromatogr. 1980, 3, 657. doi: 10.1080/01483918008060181
(65)Berthod, A.; García-álvarez-Coque, C. Micellar Liquid Chromatography; Marcel Dekker: New York, 2000.
(66)Esteve-Romero, J.; Carda-Broch, S.; Gil-Agustí, M.; Capella-Peiró, M. E, Bose, D. Trends Anal. Chem. 2005, 24, 75.
(67)Armstrong, D. W. Sep. Purif. Methods 1985, 14, 213 doi: 10.1080/03602548508068421
(68)Thomas, D. P.; Foley, J. P. J. Chromatogr. A 2007, 1149, 282 doi: 10.1016/j.chroma.2007.03.045
(69)Ruiz-ángel, M. J.; Torres-Lapasió, J. R.; García-álvarez-Coque, M. C. Anal. Chem. 2008, 80, 9705. doi: 10.1021/ac801685p
(70)Pino, V.; Yao, C.; Anderson, J. L. J. Colloid Interface Sci. 2009,333, 548. doi: 10.1016/j.jcis.2009.02.037
(71)Wang, J.; Zhang, L.; Wang, H.; Wu, C. J. Phys. Chem. B 2012,115, 4955.
(72)Rodríguez, A.; Graciani, M. M.; Moyá, M. L. Langmuir 2008,24, 12785. doi: 10.1021/la802320s
(73)Rodríguez, A.; Graciani, M. M.; Moyá, M. L. J. Colloid Interface Sci. 2009, 338, 207. doi: 10.1016/j.jcis.2009.06.005
(74)Rodriguez, J. R.; Gonzalez-Perez, A.; Del Castillo, J. L.;Czapkiewicz, J. J. Colloid Interface Sci. 2005, 250, 438.
(75)Chen, L.; Lin, S.; Huang, C.; Chen, E. Colloids Surf. A: Physicochem. Eng. Asp. 1998, 135, 175. doi: 10.1016/S0927-7757(97)00238-0
(76)Mehta, S. K.; Bhasin, K. K.; Chauhan, R.; Dham, S. Colloids Surf. A: Physicochem. Eng. Asp. 2005, 255, 153. doi: 10.1016/j.colsurfa.2004.12.038
(77)Muller, N. Langmuir 1993, 9, 96. doi: 10.1021/la00025a022
(78)Shi, L.; Li, N.; Yan, H. Langmuir 2011, 27, 1618. doi: 10.1021/la104719v
(79)Goodchild, I.; Collier, L.; Millar, S. L.; Prokěs, I.; Lord, J. C. D.;Butts, C. P. B.; Bowers, J.; Webster, J. R. P.; Heenan, R. K. J. Colloid Interface Sci. 2007, 307, 455. doi: 10.1016/j.jcis.2006.11.034
(80)Zhao, Y.; Gao, S.; Wang, J.; Tang, J. J. Phys. Chem. B 2008,112, 2031.
(81)Bhargava, B. L.; Klein, M. L. J. Phys. Chem. A 2009, 113, 1898. doi: 10.1021/jp8068865
(82)Bhargava, B. L.; Klein, M. L. J. Phys. Chem. B 2009, 113, 9499. doi: 10.1021/jp903560y
(83)Sharma, R.; Mahajan, R. K. RSC Adv. 2014, 4, 748. doi: 10.1039/C3RA42228C
(84)Singh, K.; Marangoni, D. G.; Quinn, J. G. J. Colloid Interface Sci. 2009, 335, 105. doi: 10.1016/j.jcis.2009.03.075
(85)Yuan, J.; Bai, X.; Zhao, M. Langmuir 2010, 26, 11726. doi: 10.1021/la101221z
(86)Rao, K. S.; Singh, T.; Kumar, A. Langmuir 2011, 27, 9261. doi: 10.1021/la201695a
(87)Rao, K. S.; Trivedi, T. J.; Kumar, A. J. Phys. Chem. B 2012, 116, 14363. doi: 10.1021/jp309717n
(88)Villa, C. C.; Moyano, F.; Ceolin, M.; Silber, J. J.; Falcone, R. D.;Correa, N. M. Chem. Eur. J. 2012, 18, 15598. doi: 10.1002/chem.201203246
(89)Rao, K. S.; Gehlot, P. S.; Trivedi, T. J.; Kumar, A. J. Colloid Interface Sci. 2014, 428, 267. doi: 10.1016/j.jcis.2014.04.062
(90)Chu, Z. L.; Dreiss, C. A.; Feng, Y. J. Chem. Soc. Rev. 2013, 42, 7174. doi: 10.1039/c3cs35490c
(91)Wang, H.; Zhang, L.; Wang, J.; Zhang, S. Chem. Commun. 2013,49, 5222. doi: 10.1039/c3cc41908h
(92)Shi, L.; Wei, Y.; Sun, N.; Zheng, L. Chem. Commun. 2013, 49, 11388. doi: 10.1039/c3cc45550e
(93)Du, N.; Song, R.; Zhu, X.; Hou, W.; Li, H.; Zhang, R. Chem. Commun. 2014, 50, 10573.
(94)Figueira-González, M.; Francisco, V.; García-Río, L.; Marques, E. F.; Parajó, M.; Rodríguez-Dafonte, P. J. Phys. Chem. B 2013,117, 2926. doi: 10.1021/jp3117962
(95)Rao, K. S.; Gehlot, P. S.; Gupta, H.; Drechsler, M.; Kumar, A. J. Phys. Chem. B 2015, 119, 4263. doi: 10.1021/jp512805e
(96)Wang, H.; Tan, B.; Wang, J.; Li, Z.; Zhang, S. Langmuir 2014,30, 3971. doi: 10.1021/la500030k
(97)Wang, H.; Tan, B.; Zhang, H.; Wang, J. RSC Adv. 2015, 5, 65583. doi: 10.1039/C5RA12010A
(98)Yang, J.; Wang, H.; Wang, J.; Zhang, Y.; Guo, Z. Chem. Commun. 2014, 50, 14979. doi: 10.1039/C4CC04274C
(99)Bi, Y.; Wei, H.; Hu, Q.; Xu, W.; Gong, Y.; Yu, L. Langmuir 2015, 31, 3789. doi: 10.1021/acs.langmuir.5b00107
Recent Progress in Self-Assembly of Ionic Liquid Surfactants and Its Regulation and Control in Aqueous Solutions
WANG Hui-Yong1,2LI Hong-Pei1,2CUI Guo-Kai1,2LI Zhi-Yong1,2WANG Jian-Ji1,2,*
(1Henan Key Laboratory of Green Chemistry, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, Henan Province, P. R. China;2Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, Henan Normal University, Xinxiang 453007, Henan Province, P. R. China)
Application of ionic liquid surfactants in chemical synthesis, materials preparation, and enνironmental pollution control is closely dependent on their self-assembly behaνior and aggregate structure in aqueous solution. Thus, the study of the aggregation behaνior of ionic liquid surfactants in water is of significant importance. In this reνiew, we focus our attention on the recent progress made in the regulation and control of the self-assembly behaνior of ionic liquid surfactants and related microstructure of their aggregates in aqueous solutions by alkyl chain length, cationic structure, anionic type of the ionic liquid surfactants, addition of inorganic salt and organic solνent, and enνironmental factors such as temperature, solution pH, and light. Some regularities haνe been summarized for the regulation and control of the self-assembly behaνior of ionic liquid surfactants, and the challenges to future deνelopment in this field are explained.
Ionic liquid surfactant; Aqueous solution; Self-assembly; Micelle; Vesicle; Regulation and control
O642
10.3866/PKU.WHXB201512042
Received: October 14, 2015; Revised: December 4, 2015; Published on Web: December 4, 2015.
*Corresponding author. Email: jwang@htu.cn; Tel: +86-373-3325805.
The project was supported by the National Natural Science Foundation of China (21273062, 21133009) and Key Scientific Research Project Fund of Education of Henan Province, China (15A150003, 14A150038).
国家自然科学基金(21273062, 21133009)及河南省教育厅科学技术研究重点项目(15A150003, 14A150038)资助
©Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
西安工程大学学报(2016年3期)2016-06-05
中国卫生标准管理(2015年2期)2016-01-14
现代农业(2015年3期)2015-02-28
西南军医(2015年2期)2015-01-22
应用化工(2014年1期)2014-08-16
中国药业(2014年12期)2014-06-06
食品工业科技(2014年15期)2014-03-11
郑州大学学报(理学版)(2012年4期)2012-03-25
中国洗涤用品工业(2012年8期)2012-03-20
