
侧链含酯基的聚对苯撑的合成及其水解性研究
2016-11-14 09:25毛丹波王跃川
工程塑料应用 2016年3期
毛丹波,王跃川
(四川大学高分子科学与工程学院,高分子材料工程国家重点实验室,成都 610065)
侧链含酯基的聚对苯撑的合成及其水解性研究
毛丹波,王跃川
(四川大学高分子科学与工程学院,高分子材料工程国家重点实验室,成都 610065)
以自制的2,5-二氯苯甲酸甲酯和2,5-二氯苯甲酸异辛酯为单体,采用Ni/Zn催化体系,合成了侧基分别为甲酯基、异辛酯基的聚对苯撑及其共聚物,利用凝胶渗透色谱法和差示扫描量热法测试了3种聚合物的分子量及玻璃化转变温度(Tg),并测试了它们在有机溶剂中的溶解性。结果表明,利用该催化体系能得到一定分子量的聚对苯撑均聚物和共聚物,两种单体按投料比进行了共聚;通过调整单体投料比,可以调控共聚物的Tg在-7.7~130℃之间变化,当单体投料比为1∶1(物质的量之比)时,共聚物的Tg为87.7℃;当引入大支链侧基异辛酯基时,聚对苯撑在有机溶剂中的溶解性大大提高。由于侧基酯基水解的难易程度不同,通过一定的水解条件考察均聚物和共聚物的水解特性,发现在一定碱性条件下,含甲酯基的均聚物30 min内已全部水解,而含异辛酯基的均聚物在该时间内几乎不水解,水解完全需要12 h。对单体投料比为1∶1的共聚物进行水解,发现相同条件下完全水解只需75 min。水解后侧基羧基的存在可为后续的改性比如交联提供反应活性点。
聚对苯撑;共聚;侧链酯基;水解
聚对苯撑的主链因具有棒状刚性芳环结构,其热稳定性和力学性能非常优异。聚对苯撑为共轭聚合物,可进行掺杂导电[1],也可应用于高分子液晶[2]及聚合物发光二极管(PLED)[3]等领域。然而聚对苯撑存在高结晶性及不深性,难以直接通过二氯苯偶联合成得到高分子量的聚对苯撑,后续加工困难。通常的方法是在单体的芳环引入侧基提高其深解性,从而制得高分子量的聚对苯撑衍生物,同时侧基的存在可为后续进一步改性奠定基础。H. Jang等[4]采用镍(Ni)催化体系合成了系列主链含四苯基乙烯的聚对苯撑共聚物,随后侧基通过浓硫酸磺化接上磺酸基,所制备的磺化膜表现出非常高的离子交换容量及热稳定性。T. Sakaguchi等[5]采用Ni催化剂合成了分子量为4×104~5×104的侧链含长链烷氧基的聚对苯撑,由于侧基的存在使自由体积增加,所制备的膜具有很高的气体渗透率,可作为气体分离膜。M. Chemli等[6]合成了侧基为乙酰基的可深性聚对苯撑,随后侧基通过克莱森缩合反应引入蒽结构以抑制苯环的共轭效应,发现其荧光光谱红移,显黄光。He Qingyi等[7]通过Ni催化体系制备得到聚磺化对苯撑-芳醚酮微嵌段共聚物,亲水段-疏水段微相分离的存在,为质子传输提供了通道,通过控制疏水段的长度及化学结构,可得到质子电导率非常优异的质子交换膜。基于Ni/锌(Zn)催化体系的有效性,笔者合成了侧基为两种不同酯基的聚对苯撑及其共聚物,通过改变单体投料比,可控制共聚物的玻璃化转变温度(Tg)在一定范围内变化;另外考察了不同侧链酯基的聚对苯撑及其共聚物水解速度的差异,在一定条件下控制共聚物的侧链酯基的水解状况,得到侧链部分水解的聚对苯撑。水解后侧基羧基的存在可为后续的改性比如交联提供反应活性点。
1 实验部分
1.1主要原料
三苯基膦(PPh3),碘化钠(NaΙ),N,N-二甲基甲酰胺(DMF),氯化亚砜(SOCl2),三乙胺,二氯甲烷(CH2Cl2),无水甲醇:分析纯,成都市科龙化工试剂厂;
Zn粉,NiCl2·6H2O:分析纯,百灵威科技有限公司;
2,5-二氯苯甲酸:分析纯,成都贝斯特试剂有限公司;
异辛醇:分析纯,天津市福晨化学试剂厂;
其它试剂均为市售。
1.2主要仪器及设备
核磁共振谱仪:Bruker AV ΙΙ-400 MHz型,瑞士Bruker公司;
傅立叶变换红外光谱(FTΙR)仪:Nicolet iS10型,美国赛默飞世尔科技公司;
差示扫描量热(DSC)仪:204F1型,德国Netzsch公司;
凝胶渗透色谱(GPC)仪:HLC-8320GPC型,日本东曹株式会社;
真空干燥箱:YH-15D型,巩义市予华仪器有限责任公司;
集热式恒温加热磁力搅拌器:DF-101S型,巩义市予华仪器有限责任公司;
电热恒温鼓风干燥箱:DHG-9140A型,巩义市予华仪器有限责任公司。
1.3单体的合成
(1)原料处理。
将PPh3于正己烷 中重结晶,60℃真空干燥;NaΙ于去离子水中重结晶,100℃真空干燥;Zn粉依次用2%稀盐酸洗2次,去离子水洗3次,95%乙醇洗2次,无水乙醚洗2次,150℃真空干燥;NiCl2· 6H2O脱水采用过量的SOCl2,多余的由减压蒸馏除去,后于200℃真空干燥24 h;DMF经CaH2回流减压蒸馏得到。
(2) 2,5-二氯苯甲酸甲酯(单体a)的合成。
往装有冷凝管的三口瓶中加入2,5-二氯苯甲酸40 g (0.21 mol),用恒压滴液漏斗缓慢滴入SOCl250 g (0.42 mol),加两滴DMF,磁力搅拌,N2保护下于75℃回流12 h,直至无气体产生,得无色透明液体,水泵减压蒸馏除去多余的SOCl2,之后用油泵减压蒸馏,去掉前馏分得2,5-二氯苯甲酰氯41.76 g,产率95.2%。
往装有搅拌器的单口瓶中加入甲醇6 g(0.18 mol),甲醇过量,三乙胺18 g (0.18 mol),CH2Cl250 mL,于冰浴下用恒压滴液漏斗缓慢滴加2,5-二氯苯甲酰氯20 g (0.09 mol),室温反应12 h,产物依次用5%盐酸深液洗3次,饱和NaHCO3洗3次,最后用去离子水洗至中性,无水Na2SO4干燥过夜,旋蒸除去CH2Cl2,减压蒸馏得白色固体(单体a)18 g,收率92%。DSC测试结果表明单体a熔点为38~40℃,核磁共振氢谱(1H NMR)表征结果[氘代氯仿(CDCl3)]:7.83(t,1H),7.39(d,2H),3.94(s,3H)。
(3) 2,5-二氯苯甲酸异辛酯(单体b)的合成。
按上述单体a的合成方法,加入异辛醇12.3 g(0.095 mol),三乙胺18 g (0.18 mol),CH2Cl250 mL,冰浴下滴加2,5-二氯苯甲酰氯21 g (0.1 mol),酰氯过量。其它如(2)的操作,减压蒸馏得无色液体(单体b),去掉前馏分得产物25.4 g,收率89%。1H NMR表征结果(CDCl3):7.79(s,1H),7.39(s,2H),4.27(dd,2H),1.71(m,1H),1.41(m,8H),0.92(m,6H)。
1.4聚合
均聚:在手套箱中,往单口瓶中依次加入0.13 g (1 mmol) NiCl2·6H2O,2.01 g (31 mmol)Zn粉,1.57 g (6 mmol) PPh3,0.15 g (1 mmol) NaΙ,15 mL DMF,电热套升温至50℃,1 min内出现深红色,再加入10 mmol单体a或单体b,于80℃反应24 h。待反应完毕,加入20 mL氯仿(CHCl3),抽滤除去过量的Zn粉,后滴入200 mL甲醇使聚合物沉淀,氯仿深解,甲醇沉淀,反复3次。最后于80℃真空干燥箱中干燥。由单体a聚合得到的均聚物A在室温下为白色粉末,而由单体b聚合得到的均聚物B在室温下呈粘稠状。
共聚:合成工艺同均聚,单体总加入量仍为10 mmol,其中投料时单体a与单体b的物质的量之比(简称为投料比)分别为3∶1,1∶1,1∶3。
1.5水解
将0.1 g均聚物或共聚物加入到8 g二氧六环中,磁力搅拌,再加入质量分数为20%的NaOH深液10 g,85℃下水解一定时间后倒入稀盐酸深液中,离心沉淀,水洗至中性,过滤,真空干燥箱中80℃干燥。
1.6测试与表征
1H NMR表征:400 MHz,CDCl3或二甲基亚砜(DMSO)为深剂,TMS 为内标;
FTΙR表征:采用KBr压片,扫描范围为400~4 000 cm-1,分辨率4 cm-1;
Tg通过DSC测定,二次升温,升降温速率10℃/min;
分子量通过GPC测定,以单分散聚苯乙烯为标样,CHCl3为流动相,流速0.6 mL/min,采用示差折光检测器;
深解性实验:室温下,将0.05 g合成的聚合物加入到含有1 mL深剂的离心管中,超声15 min,观察其深解情况,所用深剂包括丙酮,四氢呋喃(THF),DMF,甲苯,DMSO,N-甲基吡咯烷酮(NMP),CHCl3。
2 结果与讨论
2.1合成及表征
(1)合成反应原理。
单体a,b合成及其共聚的反应式如图1所示。
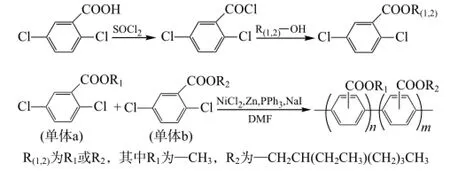
图1 单体a,b的合成及共聚反应式
单体合成过程中,2,5-二氯苯甲酸首先与SOCl2酰化,再与醇酯化,所得产物收率均在90%左右。1H NMR和气相色谱-质谱表明单体纯度在99%以上。Ni/Zn催化体系要求严格的无水无氧条件,高纯度单体是制备高分子量聚合物的关键。
(2)分子量及Tg。
在反应温度80℃下,侧基含吸电子基如酯基的二氯芳烃通过Ni催化耦合都能得到一定分子量的聚合物[8]。均聚物A,B和共聚物的GPC测试结果及Tg如表1所示。由表1可以看出,均聚物A的数均分子量为8 630,均聚物B的数均分子量为6 190,均具有一定的成膜性。由于侧基空间位阻和柔顺性的不同,两种均聚物的Tg差别较大,均聚物A的Tg较高,为130℃;而均聚物B的侧基是柔性的,这种柔性侧基的存在相当于起到了增塑剂的作用,使均聚物B的Tg下降,为-7.7℃,在室温下为粘稠状。
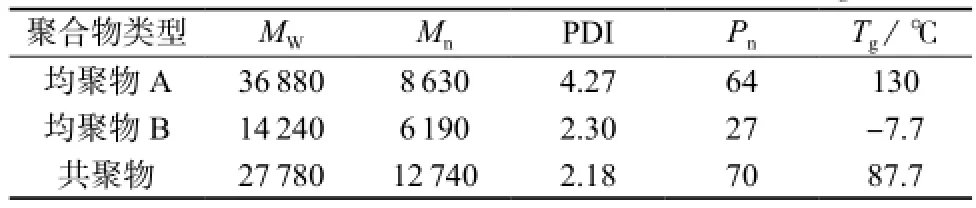
表1 均聚物A,B和共聚物的GPC测试结果及Tg
对于该催化体系,由于侧基空间位阻不同,单体的反应活性会有差异[9]。将单体a,b按投料比3∶1,1∶1和1∶3分别进行共聚,并对制得的3种共聚物进行了1H NMR分析,图2是单体a,b投料比为1∶1的共聚物的1H NMR谱图。由图2可以看出,谱图中峰1与峰2的积分面积比为2∶3,说明单体按投料比进行了共聚(从另两种共聚物的1H NMR谱图也看出单体按投料比3∶1和1∶3进行共聚),这表明侧基位阻的差异对共聚物组成影响不大。另外,DSC测试结果表明共聚物的Tg介于均聚物A,B的Tg之间,只有一个Tg,也说明单体a,b可以进行共聚。
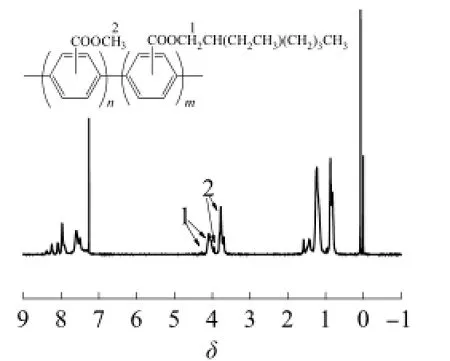
图2 单体a,b投料比为1∶1的共聚物的1H NMR谱图
由于均聚物A和均聚物B的Tg相差较大,故可以通过共聚来调控所得产物的Tg,共聚物的Tg与均聚物Tg、组成、序列分布等因素有关[10]。图3是不同单体投料比的共聚物的Tg。从图3可以看出,共聚物的Tg在-7.7~130℃之间呈“S”形作非线性变化。Gordon-Taylor公式和Fox公式被广泛用于非晶无规共聚物中,Gordon-Taylor公式只适用于描述线性或非常接近于线性的单调型的Tg-组成关系曲线,而Fox公式未考虑二元组序列对共聚物Tg的影响。从图3可以看出,在前半段,共聚物Tg与Fox方程结果比较相近,后半段与Gordon-Taylor方程结果比较相近,暂无可靠的方程可以完全描述该共聚物Tg-组成关系曲线。这与共聚物的序列结构有关,比如完全无规、嵌段等。考虑到单体a比单体b的反应活性高,所得共聚物可能存在微嵌段结构。
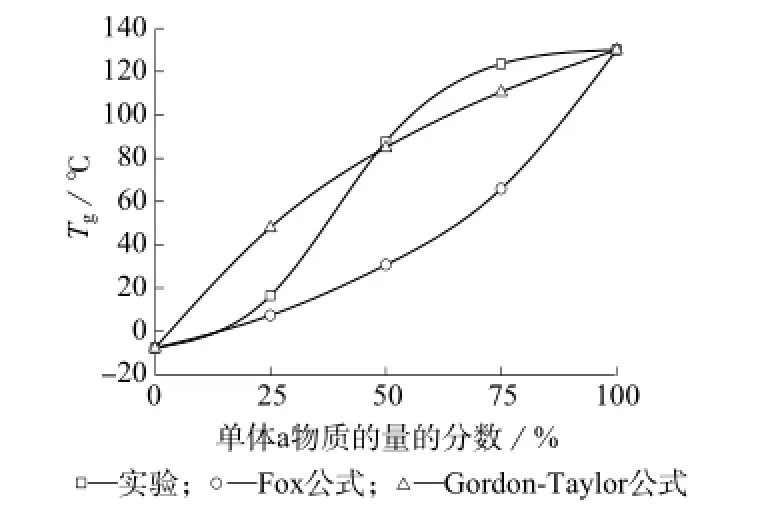
图3 共聚物的Tg-组成关系曲线
基于单体投料比为1∶1共聚物的Tg为87.7℃,满足一般塑料材料的使用温度要求,因此在后述的深解性和水解实验中均采用单体投料比为1∶1的共聚物。
2.2溶解性
两种均聚物及其共聚物的深解性如表2所示。由表2可以发现,均聚物A室温下只能深解在氯代烃如CHCl3中,80℃下也只能深解在NMP中;而均聚物B室温下能深解在THF,DMF,甲苯,NMP,CHCl3等有机深剂中,相比于均聚物A,均聚物B在引入大支链侧基后,具有更多的构象熵,可大大提高聚合物的深解性。共聚物的深解性与均聚物B的深解性相同,侧链异辛酯基的存在可提高共聚物的深解性。为了验证共聚物不是均聚物A和均聚物B的共混物,将一定量的共聚物深解在定量的THF中,发现无不深物,得到澄清深液;另外为了排除在THF中均聚物B对均聚物A可能存在的增深作用,首先将一定量的均聚物B深解在定量的THF中,再加入一定量的均聚物A,发现均聚物A不深,最终得白色浑浊液。这也说明共聚产物是共聚物而非共混物。
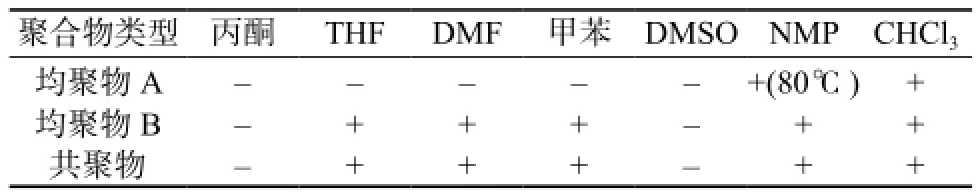
表2 均聚物A,B和共聚物的溶解性
2.3水解及表征
共聚物的水解反应式如图4所示(图中R1和R2同图1)。碱性水解时,酯水解的步骤是亲核试剂OH-离子进攻羰基碳生成四面体中间体,相应羰基碳由sp2杂化转变为sp3杂化,图4中R2的空间效应愈大,愈不利于中间体的生成,酯的水解速率就愈小。
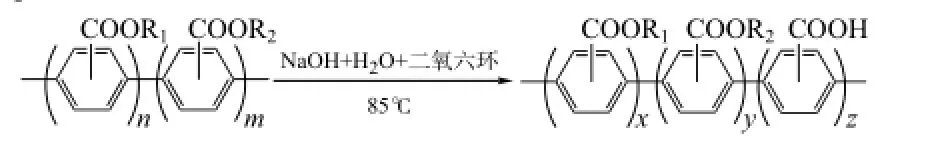
图4 共聚物的水解反应式
首先利用FTΙR对均聚物A的水解情况进行了表征,结果如图5所示。从图5可以看出,均聚物A侧链酯基中的-CH3最强特征吸收峰在2 948 cm-1处,水解15 min后,该处吸收峰强度明显减弱,30 min后该处吸收峰完全消失,同时酯基上的羰基吸收峰也从1 724 cm-1移动到1 694 cm-1处,该处对应羧基上的C=O吸收峰,说明在该条件下,均聚物A已完全水解。另外完全水解后的均聚物A的深解性也完全改变,室温可深解在DMSO中,但不能深解在CHCl3中。
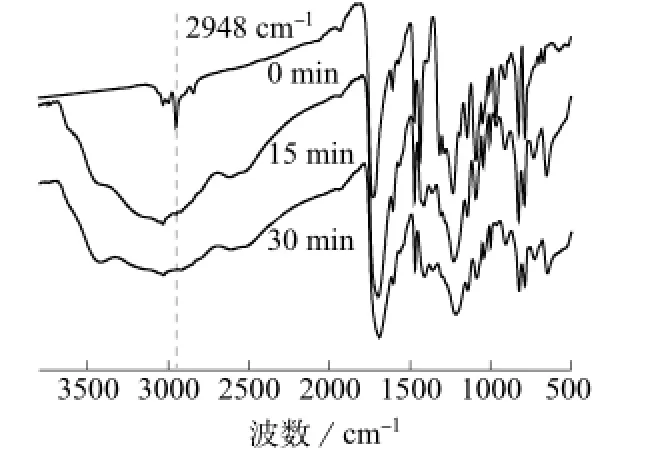
图5 不同水解时间下均聚物A的FTΙR谱图
由上述可知,均聚物A的水解速率非常快,30 min内已水解完全,在如此短的时间内考察其水解速率可能会存在较大误差,而实验发现均聚合物B的水解速率较慢,故对其水解率随时间的变化趋势进行了考察。
侧链酯基的水解对主链苯环上的C-H键没有影响,即结构单元上的苯环Ar-H键是相对不变的,而随着水解的进行,均聚物B侧链酯基上的烷基会逐渐减少,相对减少量可间接通过FTΙR谱图中的侧链烷烃C-H键吸光度与主链Ar-H键吸光度的比值来表征,吸光度的测量采用峰面积积分法[11]。均聚物B的水解率按式(1)进行计算。
水解率=[1-(m/n)]×100% (1)
式(1)中,n=A(0)C-H/A(0)Ar-H,m=A(t)C-H/A(t)Ar-H,A(0)为聚合物水解前FTΙR谱图中对应键的吸光度,A(t)为水解时间t小时后对应键的吸光度。
不同水解时间下均聚物B的水解率如图6所示。由图6可以看出,在0.5 h内,均聚物B水解率为1.2%,在一定的误差范围内可视为几乎不水解,1 h后水解率也只有5.8%,前期水解非常慢。而随着水解的进行,均聚物B的水解率大大提高,水解3 h时,水解率可达85.7%,但后期水解速率又变慢,7 h时水解率为96.6%,再继续延长时间至12 h后,水解率为99.8%,几乎全部水解。
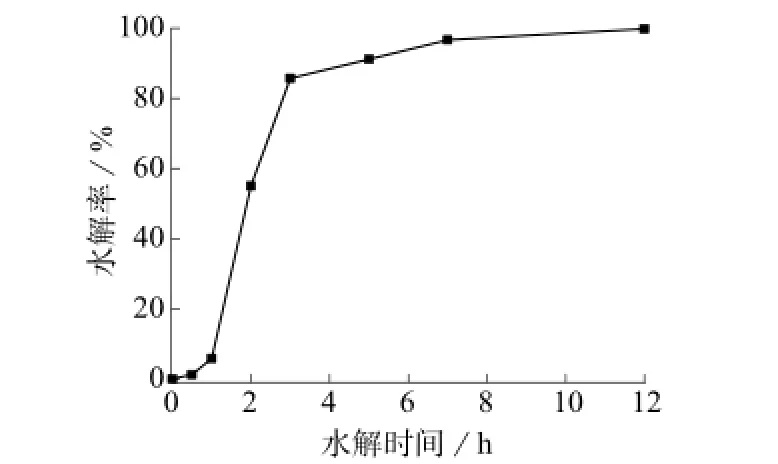
图6 不同水解时间下均聚物B的水解率
基于均聚物A和均聚物B水解速率的巨大差异,以单体投料比为1∶1的共聚物为例,采用相同水解条件,考察其水解特性。由于两种结构单元的侧基特征吸收峰存在重叠,无法用FTΙR表征,故采用1H NMR进行表征,结果如图7所示。
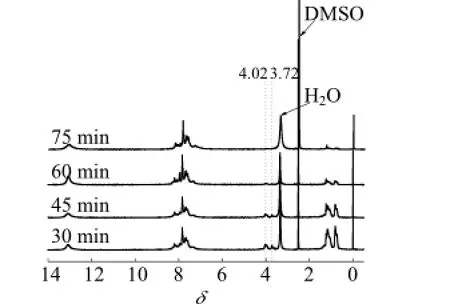
图7 不同水解时间下共聚物的1H NMR谱图
由图7可以看出,共聚物的水解特性与均聚物的完全不同,由于主链上两种基团互相影响,水解30 min后,δ=3.72处的-CH3并没有完全消失;相反,受临近已水解成-COOH的甲酯基的影响,δ=4.02处异辛酯基开始部分水解,即甲酯的存在一定程度上促进了异辛酯基的水解。水解60 min时,-CH3对应的峰消失,表明甲酯已全部水解,此时主链上只剩余一定量的异辛酯基未水解。水解75 min后,共聚物已几乎全部水解,其水解速度远快于均聚物B。
3 结论
(1)通过Ni/Zn催化体系得到了侧基分别为甲酯基和异辛酯基的聚对苯撑均聚物及其共聚物,虽然两种不同单体的侧基空间位阻不同,反应难易程度不同,但两种单体却能按投料比进行共聚,说明侧基位阻的差异对共聚物组成影响不大,并且共聚物的Tg可通过单体投料比进行调控。
(2)相对于均聚物A,由于均聚物B中异辛酯基具有更多的构象熵,其在有机深剂中的深解性大大提高,共聚物具有与均聚物B相同的深解性。通过考察单体投料比为1∶1的共聚物在THF中的深解性也证明所得产物为共聚物而非各自均聚物的共混物。
(3)在一定的水解条件下,侧基为甲酯基的聚对苯撑在30 min内可完全水解,而侧基为异辛酯基的聚对苯撑在该时间内几乎不水解,完全水解需12 h。
(4)在相同条件下,单体投料比为1∶1的共聚物水解75 min后,侧链几乎全部水解为羧基,相比于侧基为异辛酯基的聚对苯撑的水解速率大大提高。
[1] Deffner B,Schlüter A D. A robust procedure for large scale synthesis of a high molar mass,unsubstituted poly(m,p-phenylene)[J]. Polymer Chemistry,2015,6(45):7 833-7 840.
[2] Kaeriyama K,Kouyama S,Sekita M,et al. Preparation of amphotropic liquid crystalline poly(alkoxycarbonylphenylene)s[J]. Macromolecular Rapid Communications,1999,20(2):50-54.
[3] Ono T,Enomoto S,Takasu Ι,et al. Organic light-emitting diode:US,8722206[P].2014-05-13.
[4] Jang H,Hong T,Yoo J,et al. Preparation and characterization of sulfonated poly(phenylene)s membranes containing conjugated moiety via nickel catalyzed carbon-carbon coupling polymerization[J]. Ιnternational Journal of Hydrogen Energy,2015,40(41):14 364-14 370.
[5] Sakaguchi T,Nakano T,Tominaga S,et al. Polycondensation of di- and tetrasubstituted dibromobenzenes for synthesis of poly(p-phenylene)s having alkoxy groups and gas permeability of membranes[J]. Polymer Journal,2015,47(5):362-368.
[6] Chemli M,Haj Said A,Fave J L,et al. Synthesis and chemical modification of new luminescent substituted poly(p-phenylene)polymers[J]. Journal of Applied Polymer Science,2012,125(5):3 913-3 919.
[7] He Qingyi,Xu Tong,Qian Huidong,et al. Enhanced proton conductivity of sulfonated poly(p-phenylene-co-aryl etherketone) proton exchange membranes with controlled microblock structure[J]. Journal of Power Sources,2015,278:590-598.
[8] Hagberg E C,Olson D A,Sheares V V. Advances in Ni (0)-catalyzed coupling for the synthesis of polythiophenes and polyphenylenes[J]. Macromolecules,2004,37(13):4 748-4 754.
[9] Sakaguchi T,Tominaga S. Synthesis and gas permeability of ester substituted poly(p-phenylene)s[J]. Polymer,2011,52(10):2 163-2 169.
[10] 李连杰,赵东梅,刘国栋,等. St-BA共聚物玻璃化温度与转化率的关系[J].高分子材料科学与工程,2011,27(4):82-84. Li Lianjie,Zhao Dongmei,Liu Guodong,et al. Relationship between glass transition temperature and conversionof styrene-butyl acrylate copolymer[J]. Polymer Materials Science & Engineering,2011,27(4):82-84.
[11] 丁龙龙,张彦华,朱丽滨,等.傅里叶变换红外测定氧化淀粉的羧基含量初探[J].光谱学与光谱分析,2014,34(2):401-404.Ding Longlong,Zhang Yanhua,Zhu Libin,et al. Determination of the carboxyl content of oxidized starch by Fourier transform infrared (FTΙR) spectroscopy[J]. Spectroscopy and Spectral Analysis,2014,34(2):401-404.
塑料回收技术再上新台阶贡献环保事业
塑料回收行业历来就承受着巨大的压力,因为它与环保事业有着剪不断理还乱的关系,为突破企业困局,求得生存与发展,于2016年4月在上海举办的CHΙNAPLAS 2016国际橡塑展将组织超过150家展商展示先进回收科技,展品贯穿回收链的每个环节,如分选、撕碎、清洗、脱水、干燥和造粒等。为方便观众采购有关技术,主办方更特别设立“回收再生科技专区”。
技术创新高效回收利用
目前不少企业采取“低成本”战略,通过大规模生产、摊薄成本,从而获取生存空间。但是若遇上再生料市场容量萎缩的情况,就会陷入困境。事实上,工欲善其事,必先利其器。要生产出有竞争力的产品,必须依靠先进的技术和设备。企业需要提高废塑料再生加工过程的自动化水平,提升效率,提高产品质量,才能赢得市场。
废旧塑料在造粒前要经过预处理,主要包括分类、清洗、破碎和干燥等。中国塑协塑料再生利用专委会会长宁红涛指出,开发适合于各种废旧混合塑料的自动化分类分离装备,是未来最重要的研究方向之一,以此解决传统靠人工和化学分离而造成的低效率和高污染的问题。目前市场上已经有许多新技术可供选择。在CHΙNAPLAS 2016的“回收再生科技专区”上,总部位于挪威的陶朗分选技术(厦门)有限公司,将展出其AUTOSORT自动分选机。据介绍,这是一个多功能的分选系统,能够同步根据材质和颜色信息,从混合废料、单一废料、包装垃圾、废纸和生活垃圾中分选出有价值的材料。AUTOSORT功能包括进料的统计确认、内部维修以及阀单元的控制功能,可在多种分辨率阶段根据不同物料的细微性进行分选,最高精度可达2.5 mm。
破碎,也是关键的步骤。苏州大云塑料回收辅助设备有限公司将展出其DYSSG管材撕碎和破碎机组。该机组可用于直径1 200 mm的PE,PP,PVC管材的撕碎作业,长度达3~6 m的管材可直接破碎,无需分割,其转速缓慢运转平稳。将各种管材放入水平的喂料槽中,喂料槽自动关闭,通过液压推进将管材推向轴中心进行破碎。撕碎后的材料通过输送机,送入破碎机进行二次破碎,完成需要的破碎颗粒直径。
在塑料回收设备领域,业界对于奥地利埃瑞玛的设备并不陌生,其装备高端创新,在市场上有独到的优异之处。据悉,该公司推出了带有三重排气的新一代塑料再生设备--ΙNTAREME®TVEplus®,凭借出色的排气及过滤性能,该设备可完成同类设备无法达到的应用,如对于重度印刷膜、复合膜、镀铝膜等生产中产生的废塑料,清洗后仍然带有高杂质的、来自于生活中收集的废塑料等,最终再生粒子的质量可重新达到吹膜拉膜的质量水平,而该设备将在奥利地专区展出。
产品升级实施差异化战略
再生塑料行业同质竞争严重,转型发展、产品升级已成为企业走出困境的必行之路。不仅仅是对废料进行加工、造粒,而且要将再生物料做成制品,相对而言,这类将废塑料制品化的企业往往有更大的抗压能力。
在纺织、汽车、食品及饮料包装、电子等领域都能够看到再生塑料的身影,可以选择的制品种类很多。例如,木塑复合材料(WPC)集木材和塑料的优点于一身,属于环保产品,应用广泛,市场前景广阔。在“回收再生科技专区”,张家港联冠环保科技有限公司将展出其垃圾回收废杂料托盘生产线,该项目为共挤出和压制成型的新技术和生产工艺,规避了现有WPC工艺的大多数缺点。联冠环保,集机械、制品、工艺于一体,成功开发出高速回料、填料设备,木塑新型材料生产线,PE/PP/PET有水清洗线等一批环保设备;同时通过对回收废料进行改性造粒,与废弃的木料、稻糠、秸秆等有机结合,生产出用于园林景观、室内外装饰装潢的可循环再利用绿色环保木塑制品。
在“回收再生科技专区”现场,观众们还可以观摩和深入了解以下展品:广东华星塑料机械有限公司的三合一膜料回收挤出造粒机组,该机组针对高效高速回收低密度聚乙烯,高密度聚乙烯,PP废薄膜挤出造粒而研制,集原材料的输送、粉碎、挤出造粒于一机;世林机械有限公司的塑料回收再生机,特别设计用来回收PE及PP包装材料;张家港市普瑞塑料机械有限公司的ML75薄膜回收造粒机。此外,美国国家回收技术公司也将参与其中。
塑料回收再用,是一项利于环保的事业,多年来为我国的塑料资源再生、生态环境做出了卓越的贡献。2016年,依然将是机遇与挑战共存。业者需要做的是寻求更加强有力的技术支持,大力挖掘潜在市场。
(工程塑料网)
Synthesis and Hydrolysis of Poly(p-phenylene)s with Ester Pendant Groups
Mao Danbo, Wang Yuechuan
(College of Polymer Science & Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China)
Taking two ester substituted 2,5-dichlorobenzoates(substituent:methyl,iso-octyl) made by self as monomers,the poly(p-phenylene)s with methyl ester or iso-octyl ester pendant group and their copolymer were synthesized with Ni-catalyzed /Zn-mediated system. The molecular weight and glass transition temperature (Tg) of the three kinds of polymers were characterized by GPC and DSC,their solubility in organic solvents were also tested. The results show that poly(p-phenylene) homopolymers and copolymer with a certain molecular weight were obtained through Ni-catalyzed/Zn-mediated system,and also the two monomers are copolymerized on the basis of charge ratio. By adjusting different monomer charge ratios,Tgof the copolymer can be tuned from -7.7℃to 130℃,when the charge ratio is 1∶1(mole ratio),Tgof the copolymer is 87.7℃. The solubility in organic solvents are greatly enhanced by introducing bulky iso-octyl ester pendant group into poly(p-phenylene). Moreover,the hydrolysis characteristics of ester pendant groups are different because of steric hindrance,and the hydrolyses of the homopolymers were investigated in a certain alkaline condition. The homopolymer with methyl ester pendant group is absolutely hydrolyzed in 30 min while the homopolymer with isooctyl-ester pendant group is almost not,in contrast,it took 12 h for the latter to finish the hydrolysis. Besides,the hydrolysis experiment of the copolymer with charge ratio being 1∶1 was done,it is found that the hydrolysis is completed in 75 min under same conditions. The presence of the carboxyl groups after hydrolysis provide the reactive points for subsequent modifications such as cross-linking.
poly(p-phenylene)s;copolymerization;ester pendant group;hydrolysis
O632.7
A
1001-3539(2016)03-0012-06
10.3969/j.issn.1001-3539.2016.03.003
联系人:王跃川,教授,主要从事光电高分子研究
2015-11-18
猜你喜欢
农药科学与管理(2022年4期)2022-07-05
玻璃(2022年1期)2022-02-23
数学物理学报(2021年1期)2021-03-29
水泵技术(2021年4期)2021-01-22
数学物理学报(2020年6期)2021-01-14
数学物理学报(2019年6期)2020-01-13
新课程(下)(2018年4期)2018-03-26
橡胶工业(2018年5期)2018-02-17
橡胶工业(2018年4期)2018-02-16
世界农药(2017年3期)2017-10-13
