
SiC-ZrC复相陶瓷先驱体的制备与性能
2016-11-03 05:35黄传进王明存
固体火箭技术 2016年5期
李 华,黄传进,王明存
(1.天津科技大学 海洋科学与工程学院,天津 300457;2.北京航空航天大学 化学与环境学院,仿生智能界面科学与技术教育部重点实验室,北京市海淀区学院路37号,北京 100191)
SiC-ZrC复相陶瓷先驱体的制备与性能
李华1,2,黄传进2,王明存2
(1.天津科技大学 海洋科学与工程学院,天津300457;2.北京航空航天大学 化学与环境学院,仿生智能界面科学与技术教育部重点实验室,北京市海淀区学院路37号,北京100191)
以炔基聚硅烷(PEPSI)为硅源和聚锆氧烷(PNZ)为锆源,制备了SiC-ZrC复相陶瓷先驱体。SiC-ZrC先驱体通过炔基的聚合、硅氢加成和锆氧烷与硅氢键的缩合,在260~340 ℃热固化。SiC-ZrC先驱体中加入乙酰丙酮镍(NIAA),会降低先驱体的固化温度、提高陶瓷产率和促进陶瓷结晶,表明乙酰丙酮镍(NIAA)对SiC-ZrC先驱体具有催化固化和促进结晶的作用。N2气氛下,先驱体PEPSI-0.8PNZ-NIAA(PEPSI∶PNZ∶NIAA质量比为1.00∶0.80∶0.01)经1 600 ℃裂解的陶瓷产率为43%。XRD和SEM-EDS结果表明,SiC-ZrC先驱体在1 600 ℃下发生碳热还原反应,得到了纯度较高的SiC-ZrC复相陶瓷,Si、Zr和C元素在陶瓷材料中均匀分布。SiC-ZrC先驱体有望作为理想的高温陶瓷基体树脂。
SiC-ZrC;先驱体;复相陶瓷;催化固化;裂解
0 引言
目前,在航空航天领域具有重要应用价值的耐高温材料主要包括硼化物、碳化物及其复相陶瓷等[1-2]。Cf/SiC复合材料具有高强度、高可靠性和耐高温等优点,在航空航天具有广阔的应用前景。SiC陶瓷作为Cf/SiC复合材料的树脂基体,具有高强度、耐高温、耐磨耗等优异性能,但Cf/SiC复合材料在有氧环境下,最高使用温度一般低于1 600 ℃,大大限制了Cf/SiC复合材料在超高温领域的应用,故寻找提高其耐高温性能的陶瓷材料,成为科研工作者的研究重点[3-6]。
ZrC是一种熔点超过3 000 ℃的超高温陶瓷材料,具有高温力学性能高和耐气动磨耗等性能,被认为是极具有应用前景的超高温材料[7-9]。在SiC陶瓷材料中加入ZrC作为增强相,可大幅度提高材料的耐高温性、韧性和强度等[10]。SiC-ZrC陶瓷材料的制备方法有多种,如反应烧结法[11]、化学反应气相法[12]、液相渗硅法[13]和聚合物先驱体转化法[14]等,聚合物先驱体陶瓷转化法集合了分子的可设计性、对设备要求低和可低温陶瓷化等优点,其具有传统陶瓷工艺无法比拟的优势[15]。
Rangaraj L等以ZrB2和SiC粉体为起始原料,采用热压烧结工艺并引入添加剂,可制得致密度高达98%以上的ZrB2-SiC复相陶瓷[16]。李秀倩等以炭毡作为纤维增强体,采用化学气相渗透工艺,研制出低密度的C/C复合材料。结果表明,包含ZrC颗粒的SiC相双组元弥散分布在C/C复合材料基体中,且随着先驱体中有机锆含量的增加,力学性能先升后降的趋势[17]。Zimmermann等以ZrH2、B4C和Si为原料,采用反应热烧结工艺,在1 890 ℃反应制备了相对密度大于99%的ZrB2-SiC复相陶瓷[18]。以上制备陶瓷材料的方法存在制备温度高、粉体粒径大和难于加工等缺陷。花永盛等以甲基三氯硅烷、氯丙烯和四氯化锆为原料,采用电化学合成法合成含锆聚硅烷,烧结后得到以SiC为主、含少量ZrC的陶瓷[19]。Tao Cai等以Zr(OC4H9)4和聚甲基氢硅炔经1 700 ℃高温裂解得到高度结晶的ZrC和SiC组分,Zr、Si和C元素在陶瓷中均匀分布[20],此方法所得的陶瓷产率低。
本文采用炔基聚硅烷和聚锆氧烷制备了高固体含量的SiC-ZrC先驱体树脂,加工成型性好,适用于液相陶瓷先驱体转化法,可在较低温度下将有机聚合物转化为无机陶瓷,陶瓷中元素Si、Zr和C在微米级别下分布均匀,同时具有较高陶瓷产率。SiC-ZrC先驱体中加入极少量乙酰丙酮镍,研究其对先驱体固化和高温裂解过程的促进作用,并对先驱体的固化过程、热解过程和热解后陶瓷的晶相组成和微观结构进行表征。
1 实验
1.1原料
含炔基聚硅烷(PEPSI)参照文献[21]用格氏法合成,纯度100 %,液体树脂,相对分子质量Mn=900、Mw=1 500。聚锆氧烷(PNZ)参照文献[22]用乙酰丙酮对异丙醇锆进行部分配位取代,实现锆酸酯的可控制水解,纯度100%,固体树脂,相对分子质量Mn=1 500、Mw=2 000,配成70 %二甲苯溶液。乙酰丙酮镍(NIAA),纯度95%,绿色粉末,上海晶纯生化科技股份有限公司。二甲苯,分析纯,北京化工厂。
1.2SiC-ZrC先驱体的制备
本文设计了PEPSI∶PNZ质量比为1.0∶0.1、1.0∶0.5和1.0∶0.8 三个比例的先驱体树脂,分别命名为PEPSI-0.1PNZ、PEPSI-0.5PNZ和PEPSI-0.8PNZ,加入二甲苯溶解,配成质量浓度为70%的二甲苯溶液(红色透明均相)。向以上三个比例的先驱体二甲苯溶液中加入乙NIAA(其质量为PEPSI的1%),溶解呈均相透明溶液,分别命名为PEPSI-0.1PNZ-NIAA、PEPSI-0.5PNZ-NIAA和PEPSI-0.8PNZ-NIAA。
1.3SiC-ZrC先驱体的固化和高温裂解
将SiC-ZrC先驱体放在真空干燥箱中于100 ℃除去二甲苯,得到黄色树脂状固体。将先驱体树脂置于管式炉中,在N2气氛下,按照以下方案进行热固化:RT~250 ℃,升温速率5 ℃ /min,在250 ℃保温1 h。将上述固化样品置于石英坩埚中,在管式炉中N2气氛下,分别在1 000、1 400、1 600 ℃下进行裂解,升温速率为10 ℃/min,得到黑色宏观多孔块状陶瓷材料。
1.4表征
真空100 ℃处理样品进行如下表征:凝胶时间测定采用平板小刀法,在160 ℃空气条件下,对样品进行测试。红外分析(FT-IR)是采用Thermo Fisher Scientific公司生产的Nicolet Nexus-470型红外分析仪,在室温下对样品进行分析,扫描范围400~4 000 cm-1。DSC在美国 TA INSTRUMENTS公司生产的Q2000差示扫描量热仪上,以N2为测试气氛,升温速率为10 ℃/min,测试范围为RT~350 ℃。
N2气氛下,250 ℃热处理样品进行如下表征:热重分析(TGA, 型号为德国NETZSCH TG209)以N2为测试气氛,升温速率为20 ℃/min,测试范围为RT~900 ℃;以空气为测试气氛,升温速率为10 ℃/min,测试范围为RT~800 ℃。
表征高温陶瓷化后的样品:采用XRD(日本理学Rigaku D/max2500型X-射线粉末衍射仪)进行样品测试,扫描范围3 °~80 °,扫描速度为8°/min。SEM-EDS采用日本电子的JSM-7500F扫描电镜,在5.0 kV下观察陶瓷样品微观形貌,在10.0 kV Mapping模式下,进行能谱和元素分布扫描,时间为30 min。
2 结果与讨论
2.1SiC-ZrC先驱体制备原理
SiC-ZrC先驱体是由富含碳元素的PEPSI和富含氧元素的PNZ经二甲苯溶解制备的均相体系;在260~340 ℃之间,SiC-ZrC先驱体通过炔基聚合、硅氢加成和锆氧烷与硅氢键的缩合发生交联反应,形成均相固体;SiC-ZrC先驱体树脂组分及其形成杂化共聚物的原理如图1所示。
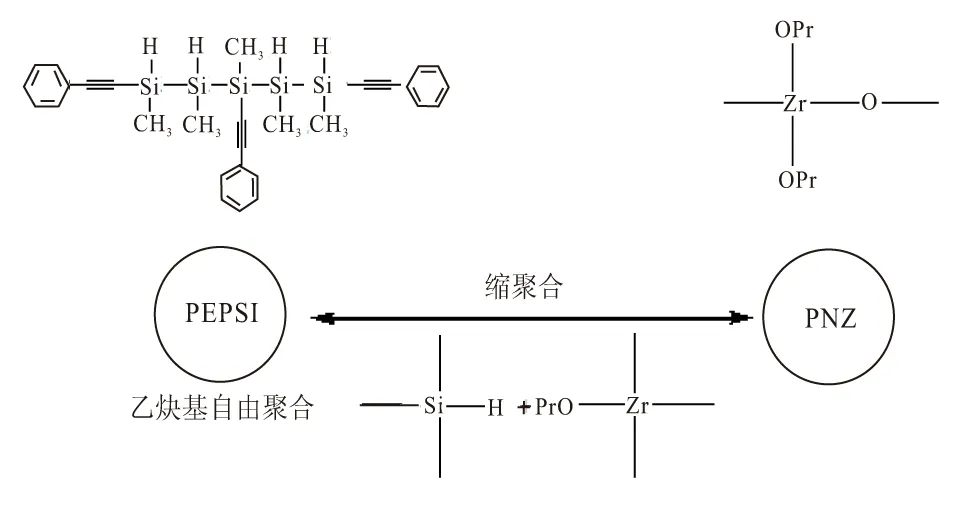
图1 SiC-ZrC先驱体树脂组分及其形成杂化共聚物的原理
N2气氛下,SiC-ZrC先驱体经1 600 ℃以上高温裂解,发生碳热还原反应(ZrO2+C→ZrC+CO↑)[19],即PEPSI中富余的C与PNZ中的O生成CO气体并释放,从而得到纯度较高的SiC-ZrC复相陶瓷。
2.2SiC-ZrC先驱体的热固化
图2为PEPSI、PNZ和PEPSI-0.8PNZ的红外谱图。从图2中可看出,PEPSI-0.8PNZ红外吸收峰包含PEPSI和PNZ各自的特征吸收峰,表明在室温下,PEPSI 与PNZ混合不发生化学反应,处于较稳定的状态,先驱体可长期稳定存放。
SiC-ZrC先驱体的热固化过程如图3所示,PEPSI-0.5PNZ固化温度较高(>260 ℃), PEPSI-0.5PNZ-NIAA固化过程分为2个阶段,固化起始温度分别为120 ℃和214 ℃,峰值温度分别为154 ℃和266 ℃,均比PEPSI-0.5PNZ固化温度明显降低,有利于先驱体的加工成型,说明NIAA对PEPSI-0.5PNZ具有催化固化的作用。
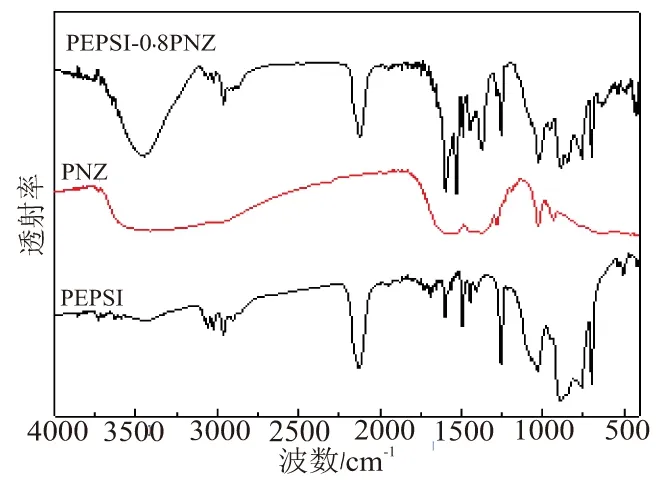
图2 PEPSI、PNZ和PEPSI-0.8PNZ的红外谱图
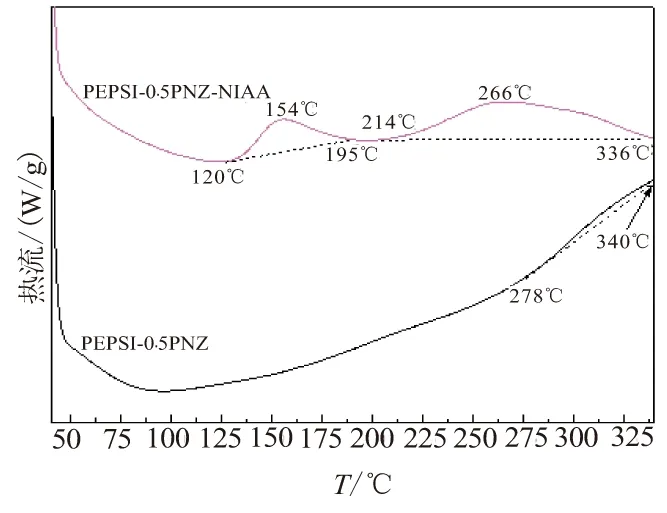
图3 PEPSI-0.5PNZ和PEPSI-0.5PNZ-NIAA样品的差热分析曲线
SiC-ZrC先驱体固化主要是炔基之间的交联反应、硅氢加成和聚锆氧烷与硅氢键之间的缩合聚合。从表1中可看出,NIAA会促使SiC-ZrC先驱体凝胶时间缩短。SiC-ZrC先驱体热固化过程会经历复杂的交联反应,图4为SiC-ZrC先驱体可能的固化机理图。交联反应主要包括炔-炔结构发生环化三聚、偶合和Diels-Alder反应形成苯环或其他交联结构[23],硅氢加成反应[24]和锆氧烷与硅氢的缩合反应[25-26]等。SiC-ZrC先驱体经过复杂的交联过程,形成均相固体,有利于先驱体经高温裂解形成均匀的SiC-ZrC复相陶瓷。
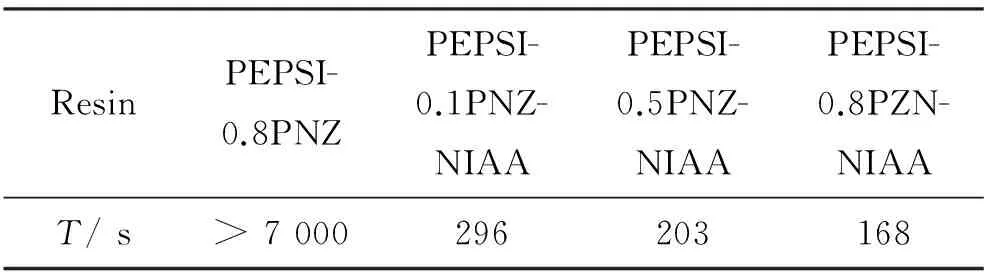
表 1 SiC-ZrC先驱体在160 ℃空气条件下的凝胶时间

图4 SiC-ZrC先驱体可能的固化机理图
2.3SiC-ZrC先驱体的高温陶瓷化
本文对SiC-ZrC先驱体进行了热失重分析,结果如图5所示。热裂解主要集中在300~650 ℃之间,该过程主要发生缩合反应,网格程度上升,主链由有机向无机方向转化,Si—H、C—H键断裂,形成无定形的三维网状结构,同时逸出碳氢化合物等气体;650~900 ℃之间失重较少,无定形三维网状结构反应结束,无机化反应进一步完成。从图5中热失重曲线中可看出,在惰性气氛下,900 ℃裂解后,SiC-ZrC先驱体加入NIAA陶瓷产率均有提高,说明NIAA对SiC-ZrC先驱体具有提高陶瓷产率的作用。
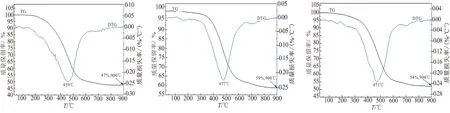
(a)PEPSI-0.1PNZ (b)PEPSI-0.1PNZ-NIAA (c)PEPSI-0.5PNZ
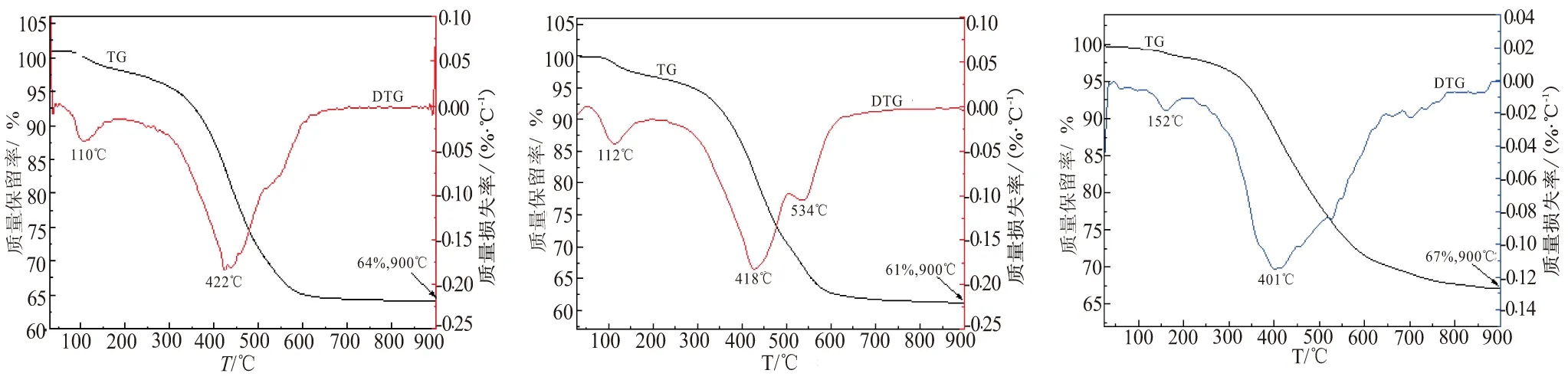
(d)PEPSI-0.5PNZ-NIAA (e)PEPSI-0.8PNZ (f)PEPSI-0.8PNZ-NIAA
SiC-ZrC先驱体经过高温裂解生成SiC-ZrC陶瓷,其裂解机理如图6所示。在惰性气氛下,随着裂解温度的不断升高,PEPSI裂解生成SiC和C,PNZ则生成ZrO2和CO、CO2等气体;裂解温度高于1 600 ℃时,发生碳热还原反应(ZrO2+C→ZrC+CO↑),得到SiC-ZrC复相陶瓷。
PEPSI→SiC+C
PNZ→ZrO2+CO(CO2)↑
ZrO2+C→ZrC+CO↑
图6SiC-ZrC陶瓷先驱体的裂解机理
Fig.6Pyrolysis mechanism of SiC-ZrC ceramic precursor
表2中,SiC-ZrC先驱体在N2气氛中不同温度下裂解的陶瓷产率可看出,在1 000 ℃和1 400 ℃下裂解,陶瓷产率接近,SiC-ZrC陶瓷主要发生的是无定形相向连续结晶相转化;1 600 ℃下,陶瓷产率较 1 400 ℃明显降低,是由于SiC-ZrC陶瓷中发生碳热还原(ZrO2+C→ZrC+CO↑),产生CO等气体导致失重。
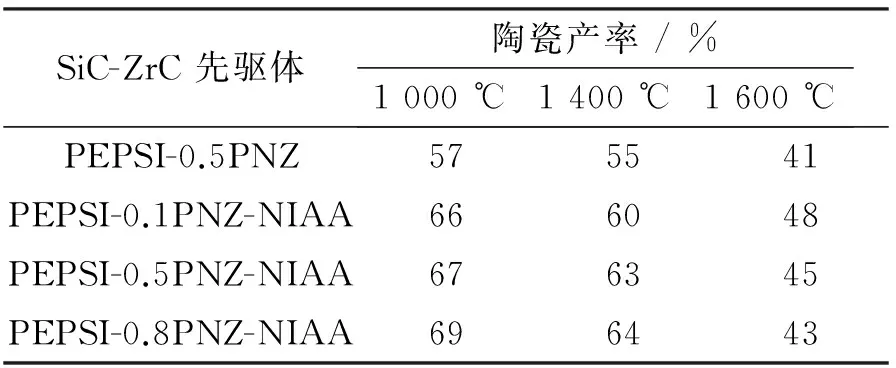
表 2 SiC-ZrC先驱体在N2气氛中不同裂解温度下的陶瓷产率(基于250 ℃处理样品)
图7中,PEPSI-0.1PNZ-NIAA、PEPSI-0.5PNZ-NIAA和PEPSI-0.8PNZ-NIAA经1 600 ℃裂解陶瓷的XRD曲线上均可看到ZrC的衍射峰(33.1 °、38.4 °、55.4 °和69.4 °)和β-SiC的衍射峰(35.6 °、41.2 °、59.9 °、71.7 °和75.3 °),无ZrO2的衍射峰,说明生成了结晶度较高的ZrC相。图8为PEPSI-0.5PNZ-NIAA在不同裂解温度下陶瓷的XRD。PEPSI-0.5PNZ-NIAA经过1 000 ℃裂解后,陶瓷的结晶主要是t-ZrO2,随着热裂解温度的升高,结晶度增强(衍射峰半峰宽变窄),ZrO2部分晶型由t-ZrO2转变为m-ZrO2,1 400 ℃裂解后的陶瓷由t-ZrO2和m-ZrO2组成,但没有出现ZrC相,经1 600 ℃发生碳热还原后,出现了较强的ZrC衍射峰,得到纯度较高的SiC-ZrC复相陶瓷。

图7 SiC-ZrC先驱体1 600 ℃裂解陶瓷的XRD曲线
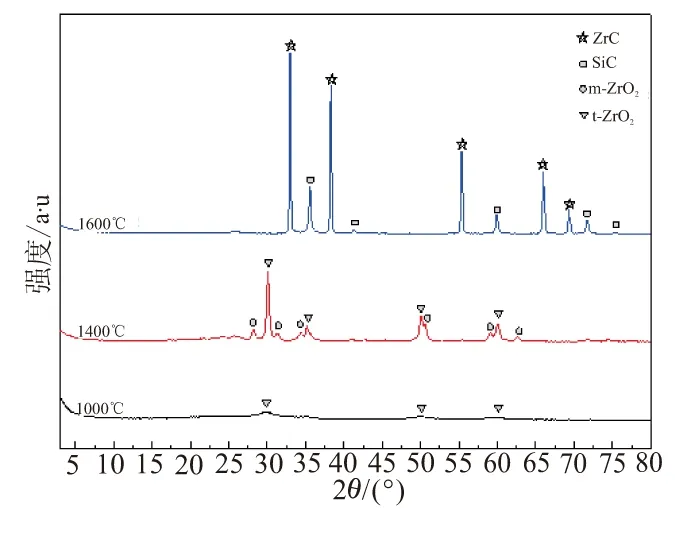
图8 PEPSI-0.5PNZ-NIAA在不同裂解温度下陶瓷的XRD曲线
图9 XRD结果可看出,PEPSI-0.8PNZ经1 600 ℃裂解后的陶瓷中有ZrO2的衍射峰,同时具有C的衍射峰(26 °),说明碳热还原未进行完全,而PEPSI-0.8PNZ-NIAA陶瓷中,却无ZrO2的衍射峰,说明NIAA具有促进陶瓷结晶的作用。
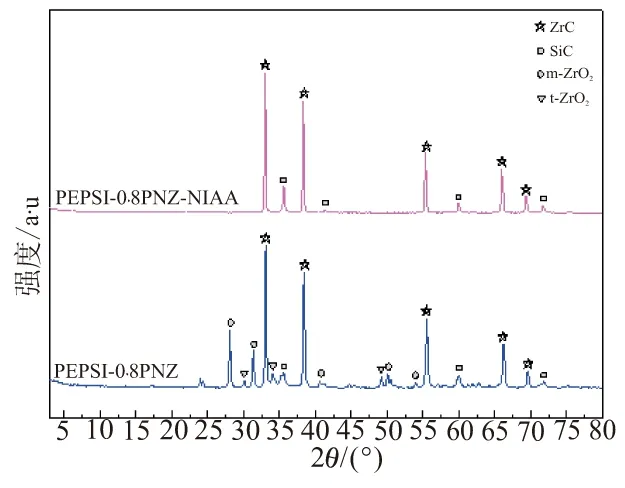
图9 SiC-ZrC复相陶瓷在1 600 ℃下陶瓷的XRD曲线
通过扫描电镜(SEM-EDS)对PEPSI-0.5PNZ-NIAA高温裂解后的陶瓷样品进行微观形貌的观察,见图10。在较低倍数(×100)下,陶瓷表面均较平整、均匀和致密。在较高倍数(×8 000)下,1 000 ℃和1 400 ℃裂解后陶瓷断面是致密的,而在1 600 ℃下的陶瓷断面则出现了纳米级孔洞,这是由于发生了碳热还原反应,产生CO等气体导致。
图11是PEPSI-0.5PNZ-NIAA陶瓷(1 600 ℃)的全视图扫描元素分布图和能谱图。

(a)×100,1 000 ℃ (b)×8 000 ,1 000 ℃

(c)×100,1 400 ℃ (d)×8 000 ,1 400 ℃

(e)×100,1 600 ℃ (f)×8 000 ,1 600 ℃
从Si、Zr和C元素分布图可看出,PEPSI-0.5PNZ-NIAA陶瓷材料中,3种元素在微米级别均匀分布。
3 结论
(1)以PEPSI和PNZ制备了SiC-ZrC复相陶瓷先驱体,先驱体在260~340 ℃热固化,交联反应机理是炔基聚合、硅氢加成和锆氧烷与硅氢键的缩合反应等。
(2)催化剂NIAA对SiC-ZrC先驱体具有催化固化和促进陶瓷化过程结晶的作用。
(3)在N2气氛下,SiC-ZrC先驱体经1 600 ℃裂解得到了SiC-ZrC复相陶瓷,陶瓷产率约为43%;在微米尺度下,经SEM-EDS-Mapping表征,Si、Zr和C元素在陶瓷样品中均匀分布。

(a) Mapping zone (b) Si mapping

(c) Zr mapping (d) C mapping
[1]Xie C,Chen M,Wei X,et al.Synthesis and microstructure of zirconium diboride formed from polymeric precursor pyrolysis[J].Journal of the American Ceramic Society,2012,95(3):866-869.
[2]Ji Z,Ye L,Tao X,et al.Synthesis of ordered mesoporous ZrC/C nanocomposite via magnesiothermic reduction at low temperature[J].Materials Letters,2012,71:88-90.
[3]Ren Z H,Jin P,Cao X M,et al.Mechanical properties and slurry erosion resistance of SiC ceramic foam/epoxy co-continuous phase composite[J].Composites Science and Technology,2015,107:129-136.
[4]Zhao S,Zhou X,Yu J,et al.SiC/SiC composite fabricated with carbon nanotube interface layer and a novel precursor LPVCS[J].Fusion Engineering and Design,2014, 89(2):131-136.
[5]Lee S P,Cho K S,Lee H U,et al. Microstructure and mechanical property of SiCf/SiC and Cf/SiC composites[C]//IOP Conference Series: Materials Science and Engineering.IOP Publishing,2011,18(16):162017.
[6]Wang H,Zhou X,Yu J,et al. Fabrication of SiCf/SiC composites by chemical vapor infiltration and vapor silicon infiltration[J].Materials Letters,2010,64(15):1691-1693.
[7]闫联生, 崔万继, 崔红, 等. 超高温抗氧化碳陶复合材料研究进展[J].宇航材料工艺,2014,44(3):6-11.
[8]Pizon D,Lucas R,Chehaide S,et al. From trimethylvinylsilane to ZrC-SiC hybrid materials[J].Journal of the European Ceramic Society,2011,31(14):2687-2690.
[9]Zhao L,Jia D,Duan X,et al.Oxidation of ZrC-30vol% SiC composite in air from low to ultrahigh temperature[J].Journal of the European Ceramic Society,2012,32(4):947-954.
[10]Zhu S, Fahrenholtz W G, Hilmas G E. Influence of silicon carbide particle size on the micro-structure and mechanical properties of zirconium diboride-silicon carbide ceramics[J].Journal of the European Ceramic Society, 2007, 27(4): 2077-2083.
[11]Katoh Y,Dong S M,Kohyama A.Thermo-mechanical properties and micro-structure of silicon carbide composites fabricated by nano-infiltrated transient eutectoid process[J]. Fusion Engineering and Design,2002,61:723-731.
[12]Kang P C, Chen G Q, Zhang B, et al. Oxidation protection of carbon fibers by a reaction sintered nanostructured SiC coating[J].Surface and Coatings Technology,2011,206(2):305-311.
[13]Patel M,Saurabh K,Prasad V V B,et al.High temperature C/C-SiC composite by liquid silicon infiltration: a literature review[J].Bulletin of Materials Science,2012,35(1):63-73.
[14]Han X Y,Cheng H F,Xing X,et al. Study on thermophysical properties of partially oxidized polymer derived SiC composites[J]. Advanced Materials Research,2012,560:86-90.
[15]Calambo P, Mera G, Riedel R, et al.Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics[J]. Journal of the American Ceramic Society,2010,93(7): 1805-1837.
[16]Rangaraj L,Divakar C,Jayaram V.Fabrication and mechanisms of densification of ZrB2-based ultra high temperature ceramics by reactive hot pressing[J]. Journal of the European Ceramic Society,2010,30(1):129-138.
[17]李秀倩,焦健,邱海鹏,等.ZrC/SiC多组元改性C/C复合材料的制备及性能研究[J].航空材料学报,2014,34(3):69-73.
[18]Zimmermann J W,Hilmas G E,Fahrenholtz W G,et al. Fabrication and properties of reactively hot pressed ZrB2-SiC ceramics[J].Journal of the European Ceramic Society,2007,27(7): 2729-2736.
[19]花永盛,陈来,李现府,等. 陶瓷先驱体含锆聚硅烷的电化学合成与表征[J].高分子学报,2011 (6):596-601.[20]Cai T,Qiu W F,Liu D,et al.Synthesis of ZrC-SiC powders by a preceramic solution route[J].Journal of the American Ceramic Society,2013,96(10): 3023-3026.
[21]Wang F,Zhang J,Huang J,et al.Synthesis and characterization of poly (dimethylsilylene ethynylenephenyleneethynylene)terminated with phenylacetylene[J]. Polymer Bulletin, 2006, 56(1): 19-26.
[22]Xie Y,Sanders T H,Speyer R F.Solution-based synthesis of submicrometer ZrB2and ZrB2-TaB2[J].J.Am.Ceram.Soc.,2008,91(5):1469-1474.
[23]刘海帆,邓诗峰,黄发荣,等.乙酰丙酮镍催化含硅芳炔树脂的固化反应行为[J]. 过程工程学报,2012,12(1):154-159.
[24]Hu H,Chen Z,Xiao J,et al.Synthesis and characterization of polysilazane precursor[J].Journal of Materials Science Letters,1999,18(16):1271-1272.
[25]Ishikawa T, Kohtoku Y, Kunagawa K. Production mechanism of polyzirconocarbo-silane using zirconium(IV) acetylacetonate and its conversion of the polymer into inorganic materials[J].Journal of Materials Science,1998,33(1):161-166.
[26]Cao F,Kim D P,Li X D,et al.Synthesis of polyaluminocarbosilane and reaction mechanism study[J].Journal of Applied Polymer Science,2002,85(13):2787-2792.
(编辑:薛永利)
Preparation and properties of SiC-ZrC multi-phase ceramic precursor
LI Hua1, 2,HUANG Chuan-jin2,WANG Ming-cun2
(1. College of Marine Science & Engineering Tianjin University of Science & Technology, Tianjin300457, China; 2. Key Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry and Environmental, Beihang University, Beijing100191, China)
A novel kind of SiC-ZrC multi-phase ceramic precursor was prepared by reactive blending of Ethynylpolysilane(PEPSI, as silicon source)and Polyzirconoxanesal(PNZ, as zirconium source). The thermal cure of SiC-ZrC precursor was realized by polymerization of ethynyl groups, hydrosilylation and condensation of zirconate with Si-H at 260~340 ℃. NIAA can reduce the curing temperature of the precursors, accelerate the pyrolysis and crystallization, and improve the ceramic yields, which shows that NIAA has an obvious catalytical effect on thermal cure and pyrolysis. For the optimized formula (PEPSI-0.8PNZ-NIAA), the monolithic ceramic was formed upon pyrolysis at 1 600 ℃ in a yield of 43% under N2atmosphere. XRD and SEM-EDS results show that: SiC-ZrC precursor is converted into almost pure SiC-ZrC multi-phase ceramic upon heat treatment at 1 600 ℃. Porous ceramic texture can occur due to the carbon thermal reduction, which generate carbon oxides. The Si, Zr and C elements are distributed uniformly in the obtained SiC-ZrC ceramic. The SiC-ZrC precursor is a promising candidate for the production of ultra-high temperature ceramics.
SiC-ZrC;precursor;composite ceramic;catalytic curing;pyrolysis
2015-06-09;
2015-07-28。
国家自然科学基金资助项目(51073053)。
李华(1989—),女,硕士,研究方向为陶瓷先驱体。E-mail:lihua19901229@163.com
王明存,副教授,从事陶瓷先驱体树脂和复合材料基体树脂的制备与应用研究。E-mail:mcwang@buaa.edu.cn
V258
A
1006-2793(2016)05-0703-06
10.7673/j.issn.1006-2793.2016.05.019
猜你喜欢
少先队活动(2022年10期)2022-12-09
中国煤炭地质(2022年10期)2022-11-23
小哥白尼(趣味科学)(2022年7期)2022-09-20
城市道桥与防洪(2022年3期)2022-05-08
陶瓷学报(2021年1期)2021-04-13
安全与环境工程(2021年2期)2021-04-02
煤炭加工与综合利用(2020年6期)2020-07-17
佛山陶瓷(2018年2期)2018-03-10
文史春秋(2017年11期)2018-01-23
优雅(2017年12期)2017-12-08
