
超顺磁Fe3O4@PDA核-壳结构纳米粒子的制备及表征
2016-10-26 03:00骆东升王新灵上海交通大学化学化工学院上海200240
功能高分子学报 2016年2期
骆东升, 王新灵(上海交通大学化学化工学院,上海200240)
超顺磁Fe3O4@PDA核-壳结构纳米粒子的制备及表征
骆东升, 王新灵
(上海交通大学化学化工学院,上海200240)
利用多元醇高温热解法和溶液氧化法制备超顺磁四氧化三铁聚多巴胺核-壳结构纳米粒子(Fe3O4@PDA)。采用X射线衍射(XRD)、透射电子显微镜(TEM)、动态光散射(DLS)、傅里叶转换红外光谱(FT-IR)和热重分析(TG)等对Fe3O4@PDA的结构、形貌和组成进行表征。采用综合物性测试系统(PPMS)对样品的磁性能进行表征。结果表明:Fe3O4@PDA的尺寸可以通过氨水与多巴胺的物质的量之比和反应时间进行调控;当Fe3O4@PDA中Fe3O4的质量分数约为5%时,具有超顺磁性,磁饱和强度为3.8 emu/g,比理论值高出36%。
超顺磁;四氧化三铁;聚多巴胺;核-壳结构
磁性四氧化三铁纳米粒子(Fe3O4)具有良好的生物相容性、生物可降解性、磁操控和磁热效应等优异性能,并能够显著缩短质子的横向弛豫时间,因此在肿瘤磁热疗、药物靶向输送、磁共振成像、物质分离等领域有着重要的应用[1-3]。当磁性Fe3O4纳米粒子的尺寸小于单磁畴的临界尺寸[4-6]时,Fe3O4纳米粒子将具有超顺磁性,即同时具有较大的磁饱和强度和被磁化后去掉磁场无剩磁[2,7]。超顺磁Fe3O4纳米粒子可以避免团聚,稳定地分散在液相中,并且可以被细胞溶酶体分解成可吸收组分,作为一种理想的纳米生物医学材料,已被美国食品药品监督管理局批准可应用于人体[8]研究中。但是由于超顺磁Fe3O4纳米粒子的尺寸较小,导致其形貌和结构不稳定,在水中或潮湿的空气中容易被氧化而腐蚀,因而在生物医药领域的应用受到限制[9-10]。因此,通过简单快速的方法对超顺磁Fe3O4纳米粒子进行表面修饰或者用聚合物等材料对其进行包覆,可以提高其性能、拓展应用范围。
二氧化硅(SiO2)和聚合物材料是常见的纳米粒子包覆材料。利用SiO2包覆纳米粒子具有包覆层的厚度易调控、复合纳米粒子分散性好的优点,但同时也存在反应条件严苛、粒子的形貌不规则和尺寸大小不均匀的缺点[11-13]。利用聚合物包覆纳米粒子的方法主要有单体聚合法和聚合物包埋法。Zhong等[14]以聚苯乙烯为包覆层利用单体聚合法制得的复合磁性微球具有较好的磁响应能力,其缺点是复合磁性微球的尺寸偏大且粒径分布较宽。Gu等[15]以聚甲基醚聚乙二醇丙烯酸酯-g-聚甲基丙烯酸(PPEGMEA-g-PMAA)为包覆层,利用聚合物包埋法制得的核-壳结构磁性微球,其形貌不规则,粒径分布宽,磁响应能力也较弱。
聚多巴胺(Polydopamine,PDA)由多巴胺(Dopamine,DA)在含氧碱性溶液[10]、酶[16]以及催化剂或强氧化剂等条件下生成具有高反应活性的醌类化合物后再经过一系列复杂的反应自聚所得[17]。PDA是一种具有黏附性、光学性能、电导性、与金属离子螯合、氧化还原性、生物相容性和生物可降解性等优异性能的聚合物材料。其丰富的羰基、氨基、羟基和儿茶酚基团具有良好的亲水性和化学反应活性,能够方便地进行活性分子、特异性基团和抗肿瘤药物的修饰和负载。例如,在碱性条件下儿茶酚基团能被氧化成醌,醌能够与巯基或氨基发生席夫碱反应或迈克尔加成反应[18]。含巯基或氨基的分子与PDA发生反应非常容易,不需要苛刻的条件或复杂的设备,在室温和碱性条件下通过简单地混合就能实现PDA的功能化。更重要的是这种交联反应发生在水中并且能够保持相当好的稳定性。以上研究表明,PDA作为一种新材料引起了科学家广泛关注和持续研究[19]。
利用PDA化学修饰Fe3O4可以将具有特异性识别功能的分子和抗癌药物负载到复合材料上,由于Fe3O4具有磁共振成像、磁靶向和磁热治疗等功能,而特异性识别功能的基团和抗癌药物具有主动靶向和化学治疗的功能,这样就能得到集诊断、多重靶向和治疗等功能于一体的新型生物医用纳米材料[20]。此外,还可以利用Fe3O4@PDA中PDA的还原性将Ag和Au等元素负载在核-壳结构纳米粒子表面应用于催化,也可以将PDA进一步碳化制得碳吸附材料,可见Fe3O4@PDA将会成为一个极具发展潜力的多功能应用平台。但目前利用PDA包覆超顺磁Fe3O4形成核-壳结构纳米粒子复合材料的研究尚鲜见报道。本文利用多元醇高温热解法合成超顺磁Fe3O4纳米粒子,利用溶液氧化法合成PDA,通过两步法制备超顺磁Fe3O4@ PDA核-壳结构纳米粒子,简单方便地制得粒子间不产生黏结、亲水性、结构稳定、尺寸均一、分散性好的超顺磁Fe3O4@PDA核-壳结构纳米粒子,并研究了反应条件与纳米粒子结构和性能之间的关系。
1 实验部分
1.1试剂
盐酸多巴胺(DA·HCl):分析纯,常州亚邦制药有限公司,溶于去离子水后DA·HCl解离得到DA,直接使用;乙酰丙酮铁(Fe(acac)3)、三乙二醇(TREG):分析纯,阿达玛斯试剂有限公司;乙酸乙酯、乙醇:分析纯,上海凌峰化学试剂有限公司;氨水(NH3·H2O):质量分数为25%~28%,上海凌峰化学试剂有限公司;磷酸二氢钾、磷酸氢二钠、邻苯二甲酸氢钾、溴化钾:分析纯,国药集团化学试剂有限公司。
1.2仪器
X射线衍射(XRD)谱:用德国Bruker公司D8 Advance型X射线衍射仪测试得到;透射电子显微镜(TEM)照片:用日本电子公司JEM-2100F型场发射透射电子显微镜得到,TEM的能量分布谱(TEM-EDS)也由此仪器测试得到;扫描电子显微镜(SEM)照片:用日本日立公司S-2150型扫描电子显微镜得到,SEM的能量分布谱(SEM-EDS)也由此仪器测试得到;动态光散射粒径分布图(DLS):用英国Malvern公司ZS90型动态光散射仪测试得到,以水为分散剂,扫描角度为90°,在25℃下测定;磁滞回线:采用美国Quantum Design公司PPMS-9T(EC-Ⅱ)型综合物性测试系统测试得到,测试温度为300 K,磁场增强步长50 Oe/s,磁场强度为-20 000~20 000 Oe;热重分析(TG):用美国TA Instruments公司Q5000IR型热重分析仪测试得到,测试的温度范围为室温~1 000℃;傅里叶转换红外光谱(FT-IR):用日本岛津公司Perkin Elmer Paragon 1000型傅里叶转换红外光谱仪测试得到。
1.3 四氧化三铁纳米粒子的制备
亲水性超顺磁四氧化三铁纳米粒子(Fe3O4)通过多元醇高温热解法合成[21]。称取707 mg Fe(acac)3,通过机械搅拌将其均匀分散到60 m L TREG中。搅拌过程中缓慢通入N2以除去反应容器和TREG中的氧气。待Fe(acac)3分散均匀后,将反应温度缓慢上升到180℃,并维持此温度45 min。紧接着增大加热功率将温度迅速升至278℃,并在此沸腾温度下继续反应45 min。反应结束后让反应体系自然冷却到室温,得到黑色液态胶体状悬浮液。将60 m L乙醇-乙酸乙酯的混合溶液(体积比为1/2)作为沉淀剂加入到反应体系中,充分搅拌后通过超速离心法分离得到Fe3O4。继续用乙醇-乙酸乙酯的混合溶液清洗3次后,将Fe3O4分散在去离子水中,并于4℃条件下保存,备用。
1.4PDA纳米粒子的制备
利用溶液氧化法制备PDA纳米粒子[10]:首先,将1.5 m L氨水、40 m L乙醇和90 m L去离子水放入250 m L的三颈圆底烧瓶中,室温下搅拌30 min;然后,将500 mg DA·HCl溶解到10 m L去离子水中形成溶液并将其缓慢滴加到反应液中,控制搅拌速率,以搅拌桨能够带动液面但不形成明显漩涡为宜,室温下反应30 h后结束反应,用高速离心法收集PDA,并用去离子水清洗3次;最后,将PDA放在45℃的真空干燥箱中干燥24 h后取出,在4℃条件下密封干燥保存。
1.5Fe3O4@PDA核-壳结构纳米粒子的制备
超顺磁Fe3O4@PDA的制备与PDA的制备方法类似。首先,将1.5 mL氨水、40 mL乙醇和90 m L去离子水放入250 mL的三颈圆底烧瓶中,室温下搅拌30 min;紧接着将Fe3O4的水相分散液滴加到上述溶液中并超声处理15 min,再将500 mg DA·HCl溶解到10 mL去离子水中形成溶液并将其缓慢滴加到反应液中,控制搅拌速率,室温下反应30 h后结束反应,用高速离心法收集产物,并用去离子水清洗3次;待清洗干净后利用强力磁铁分离得到对磁场具有快速响应能力的核-壳结构纳米粒子Fe3O4@PDA;最后,将Fe3O4@PDA放在45℃的真空干燥箱中干燥24 h后取出,在4℃条件下密封干燥保存。其合成路线图如图1所示。

图1 Fe3O4@PDA核-壳结构纳米粒子的合成路线图Fig.1 Synthesis routes of Fe3O4@PDA core-shell nanoparticles
2 结果与讨论
2.1Fe3O4的形貌和粒径分析
图2是Fe3O4的透射电子显微镜照片和动态光散射粒径分布图。从图2(a)可知,Fe3O4为形貌规则的球形,通过软件统计得到Fe3O4的粒径在8.9 nm左右。从图2(b)可知,Fe3O4的平均粒径为12 nm左右,比TEM照片的统计结果稍微偏大。这主要是因为TEM测得的是Fe3O4在干态下的粒径,DLS测得的是Fe3O4在湿态下的粒径,由于Fe3O4表面存在稳定剂TREG,使得其流体力学体积变大。根据Woo和Zhang的报道[5-6],本文制得的Fe3O4的粒径小于单磁畴的临界尺寸。
2.2Fe3O4的组成和结构分析
图3(a)是Fe3O4和TREG的红外光谱图。从图中可以看出,由于Fe3O4纳米粒子表面存在稳定剂TREG,使得Fe3O4和TREG两者的红外光谱比较相近[22],597 cm-1处的吸收峰是Fe3O4表面Fe-O键的特征吸收峰[21];3 390 cm-1处的特征峰归属于受氢键影响的羟基的伸缩振动峰,2 869 cm-1处的特征峰归属于C-H的伸缩振动峰,1 637 cm-1处的次强峰归属于C-C的伸缩振动峰,1 062 cm-1处的特征峰归属于C-O-C的伸缩振动峰。

图2 Fe3O4的透射电子显微镜照片(a)和动态光散射粒径分布图(b)Fig.2 TEM image(a)and DLS size distribution(b)of Fe3O4
图3(b)是Fe3O4的在氮气氛围下的热失重曲线,其热失重过程分为2个阶段:第1阶段在40~210℃,此阶段中失去的质量属于物理吸附的水、乙醇和乙酸乙酯,质量损失约为4%;第2阶段在210~450℃,此阶段中失去的质量属于稳定剂TREG的挥发和降解,质量损失约为8%。

图3 Fe3O4和TREG的红外光谱图(a)以及Fe3O4在氮气氛围下的热重曲线(b)Fig.3 FT-IR spectra of Fe3O4and TREG(a)and TG curve(b)of Fe3O4in N2(b)
图4(a)是Fe3O4的透射电子显微镜能谱。在0.7、6.4 ke V和7.1 ke V处的特征峰归属于Fe元素,在0.5 ke V处的特征峰归属于O元素,其余特征峰归属于测试仪器引入的元素。通过TEM-EDS测得的Fe元素和O元素的质量百分数分别为71.6%和28.4%,而理论计算得到的Fe元素和O元素的质量百分数分别为72.4%和27.6%,理论计算和测试结果基本相符。

图4 Fe3O4的透射电子显微镜能谱(a)和X射线衍射谱图(b)Fig.4 TEM-EDS spectrum(a)and XRD spectrum(b)of Fe3O4
图4(b)是Fe3O4的X射线衍射谱图。图中各个晶体特征峰与标准晶态Fe3O4卡片(JCPDS No.19-0629)中的数据相符,说明合成了结晶性好、面心立方结构的Fe3O4纳米粒子晶体。
2.3Fe3O4的分散性和磁性能
图5是Fe3O4分散在水相中30 d后的光学照片。经过30 d的静置后,分散液依旧没有出现分层,表明Fe3O4在水相中具有较好的分散性。根据Cai等[21]的报道,Fe3O4分散在水相中形成的分散液在4℃条件下储存数月之后仍能保持分散稳定。

图5 Fe3O4分散在水相中30 d的光学照片Fig.5 Photograph of Fe3O4dispersing in water for 30 d
图6是Fe3O4在300 K条件下的磁滞回线。从图中可知,Fe3O4的磁饱和强度为56.2 emu/g,具有较高的磁饱和强度。此外,Fe3O4在300 K的磁滞回线及其原点附近的局部放大图都表明:Fe3O4的磁滞回线均经过原点并且曲线基本保持重合,没有磁滞现象,矫顽力和剩磁均为零,具有超顺磁性。
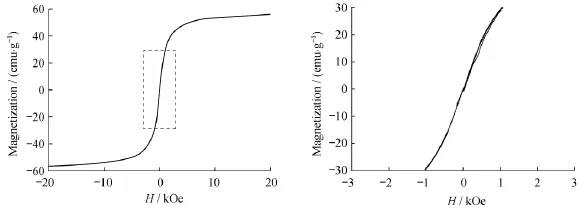
图6 Fe3O4在300 K条件下的磁滞回线(a)及其原点附近的局部放大图(b)Fig.6 Hysteresis loop of Fe3O4at 300 K(a)and enlarged view near the origin(b)
2.4PDA和Fe3O4@PDA的形貌和粒径分析
图7是PDA和Fe3O4@PDA的透射电子显微镜照片。PDA为形貌规则的纳米球,粒径小于300 nm。由于Fe3O4和PDA的电子密度不同,相应的电子束穿透能力也不同,能够明显地观察到深黑色的Fe3O4在浅灰色的PDA内部,形成以Fe3O4团簇为核的核-壳结构纳米球Fe3O4@PDA,其形貌不如PDA规则,粒径略微增大,但仍在300 nm左右。

图7 PDA(a)和Fe3O4@PDA(b)的透射电子显微镜照片Fig.7 TEM images of PDA(a)and Fe3O4@PDA(b)
图8是PDA和Fe3O4@PDA的动态光散射粒径分布图。从图8可知,PDA的粒径分布较窄,平均粒径为275 nm;Fe3O4@PDA的分布也较窄,但比PDA略宽,平均粒径为301 nm。在DA聚合包覆Fe3O4的反应过程中,Fe3O4会形成团簇,这些团聚体的形态不稳定,有的呈球状,有的呈棒状。DA自聚形成的PDA外壳包覆在Fe3O4内核外,内核的不规则性会导致Fe3O4@PDA纳米球尺寸比相同反应条件下的PDA纳米球要大,形貌规整性下降,粒径分布变宽。

图8 PDA(a)和Fe3O4@PDA(b)的动态光散射粒径分布图Fig.8 DLS size distributions of PDA(a)and Fe3O4@PDA(b)
2.5PDA和Fe3O4@PDA的组成和结构分析
图9是PDA和Fe3O4@PDA的红外光谱图。由于Fe3O4在Fe3O4@PDA中的含量较少且PDA具有较强的红外吸收,导致PDA和Fe3O4@PDA的红外吸收光谱比较相似,主要呈现PDA的特征吸收峰:3 376、3 231 cm-1处的吸收峰归属于O-H和N-H的伸缩振动;1 626、1 286 cm-1处的吸收峰归属于C=O和C-O的伸缩振动;1 511 cm-1处的吸收峰归属于C=N和C=C。

图9 PDA和Fe3O4@PDA的红外光谱图Fig.9 FT-IR spectra of PDA and Fe3O4@PDA
图10(a)为PDA和Fe3O4@PDA在氮气氛围下的热失重曲线,两者比较相似:热失重第1阶段的温度范围是40~160℃,质量损失属于PDA表面物理吸附的水分,其中PDA的质量损失约为6%,Fe3O4@PDA的质量损失约为9%;热失重第2阶段的温度从160℃开始,主要为PDA的降解,当温度上升到800℃时,PDA和 Fe3O4@PDA仍旧分别保留45%和48%的质量,表明制备得到的PDA和Fe3O4@PDA均具有较好的热稳定性。

图10 PDA和Fe3O4@PDA分别在氮气(a)和氧气(b)氛围下的热失重曲线Fig.10 TG curves of PDA and Fe3O4@PDA in N2(a)and O2(b)
图10(b)为Fe3O4@PDA在氧气氛围下的热失重曲线。从图中可知,当温度上升到600℃后,Fe3O4@PDA的热失重曲线保持平稳,剩余的质量百分比约为5%。在氧气氛围下高温煅烧,PDA完全分解,剩余的质量属于由Fe3O4氧化得到的Fe2O3。
图11(a)是PDA扫描电子显微镜能谱。通过比较Fe3O4、PDA和Fe3O4@PDA的元素组成可知,PDA仅含有C、N、O元素。图11(b)是Fe3O4@PDA的透射电子显微镜能谱。由图11(b)可知,Fe3O4@PDA中同时出现了分别属于Fe3O4和PDA的元素(C、O、Fe),其余特征峰归属于测试仪器引入的元素。

图11 PDA的扫描电子显微镜能谱(a)和Fe3O4@PDA的透射电子显微镜能谱(b)Fig.11 SEM-EDS pattern of PDA(a)and TEM-EDS pattern of Fe3O4@PDA(b)
图12是PDA和Fe3O4@PDA的X射线衍射谱图。如图12所示,PDA在23°和40°附近出现宽而强的特征峰。Fe3O4@PDA的谱图中出现归属于PDA的特征峰,23°处的特征峰移至25°并且特征峰变宽;30°、35°、43°、53°、57°和63°处出现归属于Fe3O4的特征峰,特征峰的位置几乎不发生变化。Fe3O4@PDA的谱图中同时出现PDA和Fe3O4的特征峰,说明在制备核-壳结构纳米粒子的过程中Fe3O4的结构没有被破坏,达到了利用PDA修饰并保护Fe3O4的目的。

图12 PDA(a)和Fe3O4@PDA(b)的X射线衍射谱图Fig.12 XRD patterns of PDA(a)and Fe3O4@PDA(b)
2.6纳米粒子的尺寸调控
图13(a)是PDA的粒径与n(NH4OH)/n(DA)之间的关系。从图13(a)可知,随着n(NH4OH)/n(DA)的增加,PDA的粒径不断减小。当n(NH4OH)/n(DA)较小时,通过增加氨水能够显著减小PDA的粒径,随着n(NH4OH)/n(DA)不断增大,其对PDA的粒径的影响逐渐减小。当n(NH4OH)/n(DA)在11左右时,可以制得粒径在250 nm左右的纳米粒子,与Ai等[10]报道的结果相符。
图13(b)是PDA的粒径与反应时间的关系。DA在碱性条件下能够迅速发生聚合,从宏观上看颜色快速从黄色透明变为深棕色,15 min之后变为黑色不透明。从图13(b)可知,反应3 h后PDA的粒径就达到了150 nm左右,继续反应27 h,PDA的粒径达到265 nm左右。由此可知,纳米粒子在反应前期尺寸迅速增大,之后的时间里纳米粒子的尺寸增大放缓,这主要是DA转化率的增加和PDA纳米球的形貌变得更加规则、粒径分布变窄所致。由于Fe3O4@PDA的尺寸也主要取决于PDA壳层的厚度,因此通过DA聚合时的反应条件来控制纳米粒子尺寸的方法同样适用于Fe3O4@PDA的尺寸调节。
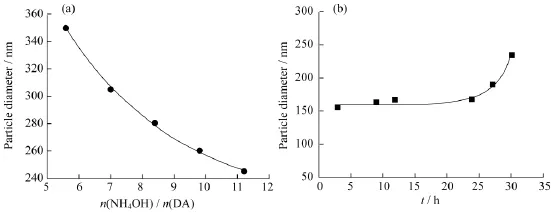
图13 PDA的粒径与氨水/多巴胺物质的量之比(a)和反应时间(b)的关系Fig.13 Diameters of PDA with different n(NH4OH)/n(DA)(a)and reaction time(b)
2.7Fe3O4@PDA的分散性和磁性能
图14是Fe3O4@PDA分散在不同p H的PBS溶液中静置1 h后的光学照片。Fe3O4@PDA在酸性和弱碱性的PBS溶液中都保持了较好的分散稳定性,分散均匀,没有产生明显的沉淀。PDA表面有大量的酚羟基,Ho等[23]报道,当环境的p H在3以上时,PDA表面将携带负电荷,形成相互排斥的静电作用,有利于Fe3O4@PDA在水相中的分散稳定性。

图14 Fe3O4@PDA分散在不同p H的PBS溶液中的光学照片Fig.14 Photographs of Fe3O4@PDA dispersing in PBS solution with different p H values
图15是Fe3O4@PDA在300 K下的磁滞回线。从图中可知,Fe3O4@ PDA的磁饱和强度为3.8 emu/g,而根据热失重分析可知Fe3O4@PDA中Fe3O4的质量分数约为5%。

图15 Fe3O4@PDA在300 K下的磁滞回线(a)及其原点附近的局部放大图(b)Fig.15 Hysteresis loop of Fe3O4@PDA at 300 K(a)and enlarged view near the origin(b)
由“相同磁性材料的磁饱和强度与其质量成正比”的关系计算得到Fe3O4@PDA的理论磁饱和强度为2.8 emu/g,所以实际测试得到的结果比理论值高出36%。这种差别出现的原因是,当超顺磁Fe3O4纳米粒子之间的距离小于其单畴尺寸时,相邻纳米粒子之间会产生电磁耦合效应,其结果是材料的磁饱和强度增大;不同数量的超顺磁Fe3O4纳米粒子聚集状态导致磁饱和强度增大的倍数也会不同。上述特征为核-壳结构超顺磁Fe3O4纳米粒子的应用创造了有利条件:只需较小的纳米粒子浓度即可满足应用所需要的性能要求。
图16是Fe3O4@PDA在磁场作用下的行为。在没有外界磁场作用时,Fe3O4@PDA能均匀地分散在水相中,并保持较好的稳定性;当存在外界磁场时,Fe3O4@PDA能够迅速地做出响应,在30 min内便聚集到磁铁一侧;当Fe3O4@PDA聚集在磁铁一侧后移去磁铁并振荡样品瓶,Fe3O4@PDA便能够重新均匀稳定地分散。

图16 Fe3O4@PDA在磁场作用下的行为Fig.16 Behavior of Fe3O4@PDA under an external magnetic field
3 结 论
(1)利用多元醇高温热解法和溶液氧化法制备以超顺磁四氧化三铁纳米粒子为核、以聚多巴胺为壳的核-壳结构纳米粒子Fe3O4@PDA。
(2)Fe3O4@PDA纳米粒子的尺寸为300 nm左右,大小可以通过氨水与多巴胺的物质的量之比和反应时间进行调控,Fe3O4的质量分数约为5%。
(3)Fe3O4@PDA纳米粒子具有超顺磁性、较好的水相分散性和磁响应性,磁饱和强度为3.8 emu/g,比理论值高出36%。
[1] LAURENT S,FORGE D,PORT M,et al.Magnetic iron oxide nanoparticles:Synthesis,stabilization,vectorization,physicochemical characterizations,and biological applications[J].Chemical Reviews,2008,108(6):2064-2110.
[2] LEE N,HYEON T.Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents[J].Chemical Society Reviews,2012,41(7):2575-2589.
[3] REDDY L H,ARIAS J L,NICOLAS J,et al.Magnetic nanoparticles:Design and characterization,toxicity andbiocompatibility,pharmaceutical and biomedical applications[J].Chemical Reviews,2012,112(11):5818-5878.
[4] ZHENG Y,CHENG Y,BAO F,et al.Synthesis and magnetic properties of Fe3O4nanoparticles[J].Materials Research Bulletin,2006,41(3):525-529.
[5] WOO K,HONG J,CHOI S,et al.Easy synthesis and magnetic properties of iron oxide nanoparticles[J].Chemistry of Materials,2004,16(14):2814-2818.
[6] ZHANG L,DOU Y H,GU H C.Sterically induced shape control of magnetite nanoparticles[J].Journal of Crystal Growth,2006,296(2):221-226.
[7] ROSEN J E,CHAN L,SHIEH D B,et al.Iron oxide nanoparticles for targeted cancer imaging and diagnostics[J]. Nanomedicine:Nanotechnology,Biology and Medicine,2012,8(3):275-290.
[8] JUNG C W,JACOBS P.Physical and chemical properties of superparamagnetic iron oxide MR contrast agents:Ferumoxides,ferumoxtran,ferumoxsil[J].Magnetic Resonance Imaging,1995,13(5):661-674.
[9] CHEONG S,FERGUSON P,FEINDEL K W,et al.Simple synthesis andfunctionalization of iron nanoparticles for magnetic resonance imaging[J].Angewandte Chemie,2011,123(18):4292-4295.
[10] AI K,LIU Y,RUAN C,et al.Sp2 C-dominant N-doped carbon sub-micrometer spheres with a tunable size:A versatile platform for highly efficient oxygen-reduction catalysts[J].Advanced Materials,2013,25(7):998-1003.
[11] DENG Y,QI D,DENG C,et al.Superparamagnetic high-magnetization microspheres with an Fe3O4@SiO2core and perpendicularly aligned mesoporous SiO2shell for removal of microcystins[J].Journal of the American Chemical Society,2008,130(1):28-29.
[12] DU G H,LIU Z,XIA X,et al.Characterization and application of Fe3O4/SiO2nanocomposites[J].Journal of Sol-gel Science and Technology,2006,39(3):285-291.
[13] ZHU L,PAN D,DING L,et al.Mixed hemimicelles SPE based on CTAB-coated Fe3O4/SiO2NPs for the determination of herbal bioactive constituents from biological samples[J].Talanta,2010,80(5):1873-1880.
[14] ZHONG W,LIU P,SHI H G,et al.Ferroferric oxide/polystyrene(Fe3O4/PS)superparamagnetic nanocomposite via facile in situ bulk radical polymerization[J].Express Polym Lett,2010,4(3):183-187.
[15] GU L,SHEN Z,FENG C,et al.Synthesis of PPEGMEA-g-PMAA densely grafted doublehydrophilic copolymer and its use as a template for the preparation of size-controlled superparamagnetic Fe3O4/polymer nano-composites[J].Journal of Materials Chemistry,2008,18(36):4332-4340.
[16] KOBAYASHI S,MAKINO A.Enzymatic polymer synthesis:An opportunity for green polymer chemistry[J].Chemical Reviews,2009,109(11):5288-5353.
[17] DELLA VECCHIA N F,AVOLIO R,ALFÈM,et al.Building-block diversity in polydopamine underpins a multifunctional eumelanin-type platform tunable through a quinone control point[J].Advanced Functional Materials,2013,23(10):1331-1340.
[18] XU L Q,YANG W J,NEOH K G,et al.Dopamine-induced reduction and functionalization of graphene oxide nanosheets [J].Macromolecules,2010,43(20):8336-8339.
[19] LIU Y,AI K,LU L.Polydopamine and its derivative materials:Synthesis and promising applications in energy,environmental,and biomedical fields[J].Chemical Reviews,2014,114(9):5057-5115.
[20] HU M,MI B.Enabling graphene oxide nanosheets as water separationmembranes[J].Environmental Science& Technology,2013,47(8):3715-3723.
[21] CAI W,WAN J.Facile synthesis of superparamagnetic magnetite nanoparticles in liquid polyols[J].Journal of Colloid and Interface Science,2007,305(2):366-370.
[22] WAN J,CAI W,MENG X,et al.Monodisperse water-soluble magnetite nanoparticles prepared by polyol process for highperformance magnetic resonance imaging[J].Chemical Communications,2007(47):5004-5006.
[23] HO C C,DING S J.The p H-controlled nanoparticles size of polydopamine for anti-cancer drug delivery[J].Journal of Materials Science:Materials in Medicine,2013,24(10):2381-2390.
Synthesis and Characterization of Superparamagnetic Fe3O4@PDA Core-Shell Structure Nanoparticles
LUO Dong-sheng, WANG Xin-ling
(School of Chemistry and Chemical Engineering,Shanghai Jiaotong University,Shanghai 200240,China)
Superparamagnetic Fe3O4@PDA core-shell structure nanoparticles were synthesized via polyol thermo decomposition and solution oxidation.The structure,morphology and component were characterized by X-Ray Diffraction(XRD),Transmission Electron Microscopy(TEM),Fourier Transform Infrared Spectroscopy(FT-IR),and Thermogravimetric Analysis(TG),etc.The magnetic properties of the samples were measured using physical properties measurement system(PPMS).Results showed that the size of Fe3O4@PDA could be controlled by adjusting the molar ratio of ammonia to DA and reaction time.When the mass fraction of Fe3O4in Fe3O4@PDA was about 5%,Fe3O4@PDA was superparamagnetic and the saturation magnetization was 3.8 emu/g,which was 36%higher than the theoretical value.
superparamagnetic;Fe3O4;polydopamine;core-shell structure
O63
A
1008-9357(2016)02-0172-010DOI: 10.14133/j.cnki.1008-9357.2016.02.005
2016-02-29
国家自然科学基金(20974061)
骆东升(1991-),男,浙江绍兴人,硕士生,主要从事生物医用材料的研究。E-mail:don.dongsheng.luo@gmail.com
王新灵,E-mail:xlwang@sjtu.edu.cn
猜你喜欢
中华实验眼科杂志(2022年1期)2022-11-15
昆明医科大学学报(2022年1期)2022-02-28
中国食用菌(2021年10期)2021-11-04
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
浙江工业大学学报(2017年5期)2018-01-22
北京航空航天大学学报(2017年2期)2017-11-24
科教导刊(2016年36期)2017-03-30
江苏农业科学(2015年11期)2016-01-27
天津科技大学学报(2015年4期)2015-04-16
