
熔融制样-X射线荧光光谱法同时测定铀钼矿中主次成分*
2016-10-25 08:16:18柳金良张鑫宋茂生
化学分析计量 2016年5期
柳金良,张鑫,宋茂生
(核工业230研究所,长沙 410007)
熔融制样-X射线荧光光谱法同时测定铀钼矿中主次成分*
柳金良,张鑫,宋茂生
(核工业230研究所,长沙 410007)
采用混合熔剂熔融制样,建立了同时测定铀钼矿中U,Mo,SiO2,Fe2O3,Al2O3等的X射线荧光光谱法。以Li2B4O7-LiBO2作为熔融试剂,NH4NO3作为样品的氧化剂,样品与熔剂的质量比为1∶10。除使用铀矿石国家标准物质外,主要选取人工混合校准样品及历年实验室比对的铀钼矿校准样品绘制成工作曲线。采用理论系数法校正样品的基体效应,用Br Kα,Zr Kα,U Lα,Ba Lα,Zn Kα谱线扣除相应元素的谱线重叠干扰。该方法各组分测定结果的相对标准偏差均小于2.0%(n=10);用标准物质验证方法的准确度,测定值相对误差在0.00~8.00%之间,与标准值基本吻合。该法应用于生产实验中,可以满足对铀钼矿准确、快速的测量要求。
X射线荧光光谱;熔融制样;铀钼矿;主次成分
自然界中约有39%的铀矿床伴生或共生其他金属元素[1],其中铀钼矿是一种比较常见的铀伴生矿资源。随着目前国内核电站数量和铀需求量的增加,开发利用铀矿及其伴生矿资源日趋紧迫。文献[2-7]表明,从技术与经济上对铀钼矿的开采选冶以及分离回收都是可行的。因此准确测量铀、钼含量对铀钼矿的开发利用有直接影响,而测量其它组分含量对选矿工艺和全面认识铀钼矿亦有指导意义。目前,测量铀钼矿石中各组分含量主要以化学法为主[8-10],这些方法样品前处理麻烦,多以单成分测定为主,不能同时测定多种成分。虽然文献[11]报道了关于ICP-AES测定钼矿石成分的方法,但是不能满足全部主次成分的测量,而且高含量元素测量误差较大。
近年来,X射线荧光光谱法发展迅速,用该方法同时测量矿石中主次成分已经逐步得到人们的认可和推广。杨小丽等[12]对X射线荧光光谱测量钼矿石主次成分的方法进行了报道;田文辉等[13]探讨了用能量色散X射线荧光光谱仪测定钼矿石中钼铅铁铜的方法。针对X荧光光谱法测定铀钼矿中主次成分的文献则较少。
笔者用Li2B4O7-LiBO2作为熔融试剂,熔样质量比为1∶10,为防止铂金坩埚被腐蚀,以硝酸铵为氧化剂,确定预氧化温度为550℃,制成玻璃片。除使用一级铀矿石标准物质外,重点选取自制的及历年实验室比对的铀钼矿校准样品制作工作曲线,用XRF法对铀钼矿中U,Mo,SiO2,Fe2O3,Al2O3等12种组分进行测定,取得了满意的实验结果。
1 实验部分
1.1 主要仪器与试剂
X射线荧光光谱仪:帕纳科AxiosMAX 波长色散型,最大激发电压为60 kV,最大电流为160 mA,最大功率为4.0 kW,附带SSTmAX超尖锐端窗铑靶X光管、孔内径32 mm的进样杯、SuperQ5.1A软件,荷兰PANalytical公司;
熔样机:TNRY-01C型,洛阳特耐仪器有限责任公司;
三边铂黄坩埚:底内径34 mm,含Au 5%,Pt 95%,常宏贵金属有限公司;
NH4NO3,NH4Br:分析纯,天津市博迪化工有限公司;
铀矿石标准物质:GBW 04101~GBW 04105,核工业北京化工冶金研究院;
铀矿石标准物质:GBW 04117~GBW 04122,核工业北京地质研究院。
1.2 分析条件
各元素的测量条件见表1。

表1 待测元素的测量条件
1.3 熔融片的制备
分别称取烘干过的样品0.700 0 g和7.000 g Li2B4O7-LiBO2混合熔剂(质量比为67∶33),置于特制的玻璃杯中。搅拌混合均匀后,转移至预先加有固体NH4NO3的铂黄坩埚中。在550℃马弗炉中预氧化8 min,取出冷却,加入5 d 20% NH4Br,置于1 100℃熔样机中,预熔2 min,熔融15 min。熔融过程中,自动摇摆并赶尽气泡,熔样机停止摇摆后,用不锈钢坩埚钳小心夹出铂黄坩埚,依次置于瓷板上,自然冷却脱模,在玻璃片上贴好标签,待测。
1.4 校准样品的选取
选择铀矿石国家一级标准物质(编号分别为GBW 04101 ~GBW 04105,GBW 04117~GBW 04122)作为校准样品,以研磨配制的标准物质RPUMo 01~RPUMo 07,实验室比对的标准物质BDUMo 01~BDUMo 08作为补充,校准样品各组分含量范围见表2。

表2 校准样品各组分含量范围
1.5 基体效应和谱线重叠的干扰校正
熔融制样是将样品经高温熔融于混合熔剂中,可以消除样品间成分、密度和粒度的不均匀性,以及矿物、粒度效应,同时可降低各组分间的基体效应。由于样品中组分含量变化大,仍然需要继续进行基体校正[14]。笔者采用消去烧失量-理论α系数法校正基体效应,用Br Kα,Zr Kα,U Lα,Ba Lα,Zn Kα扣除相应元素的谱线重叠干扰。仪器所用的综合数学校正公式见式(1)[15]:

式中:Ci——未知样品中分析元素i的含量;
Di——分析元素i的校准曲线的截矩;
Lim——干扰元素m对分析元素i的谱线重叠干扰校正系数;
Zm——干扰元素m的含量或计数率;
Ei——分析元素i校准曲线的斜率;
Ri——分析元素i的计数率(或与内标线强度的比值);
αij,βij,δij,γijk——基体校正因子;
Zj,Zk——共存元素j,k的含量或计数率;
n——共存元素的数目。
2 结果与讨论
2.1 熔样比例
设计试样与Li2B4O7-LiBO2混合熔剂质量比分别为1∶5,1∶10,1∶20,固定其它试验条件,熔融制备玻璃样片。多次试验结果显示,熔样比例为1∶5时,存在容易裂片、玻璃片中有白点且不透明的现象;熔样比例为1∶10,1∶20时,制得玻璃片均匀透明且脱模容易。考虑到低含量组分U,Mo,Mn和轻元素Na,Mg的测量误差,选择试样与熔剂的质量比为1∶10作为本实验的熔样比例。
2.2 样品预氧化
地质样品中一般存在硫、磷、有机质等还原性物质,在高温熔样过程中若不对样品进行预氧化,铂黄坩埚容易被腐蚀,甚至脆化开裂。氧化剂一般可以选择固体NH4NO3,LiNO3。相对于固体LiNO3,固体NH4NO3灼烧后残渣量可忽略不计,加入的量不需精确控制,制样的重现性更好,因此本实验选择固体NH4NO3作为氧化剂。氧化剂加入方式上,选择在坩埚底部铺一层固体NH4NO3,然后再倒入样品和熔剂的混合物,这样可以确保样品与坩埚接触面的还原性物质能被彻底氧化,效果较佳;考虑到固体NH4NO3在高温下反应剧烈,多次试验表明选择预氧化温度为550℃时样品不会出现喷溅现象。
2.3 校准样品的确定
X射线荧光光谱法是一种相对分析技术,其准确性很大程度上依赖于一套高质量的校准样品[16],市面上有一套铀矿石国家标准物质,但各组分存在含量梯度不够、含量范围较窄的缺点,特别对U,Mo的影响更加显著,因此需要用其它的校准样品作补充。笔者以GBW 04101、光谱纯MoO3为基准,通过和一些国家标准物质研磨混合,制备了一套人工合成的校准样品RPUMo 01~RPUMo 07;另外,搜集了一套历年来至少10家以上实验室间比对过的铀钼矿校准样品BDUMo 01~BDUMo 08。以上3套校准样品不仅解决了各组分的含量梯度和范围问题,而且由工作曲线中各组分的RMS和K值(表3)可知,上述校准样品的选择可行。

表3 工作曲线中各组分的RMS和K值
2.4 方法检出限和测定下限
X射线荧光光谱法中各组分的检出限按式(2)计算[17],以3倍检出限作为测定下限,结果见表4。

式中:m——单位含量的计数率(斜率);
Ib——背景计数率;
t——峰值和背景计数时间。

表4 方法检出限 μg/g
2.5 方法精密度和准确度
选择铀钼矿管理样M1,经本法制备10个玻璃片,按表1的测量条件上机测试,所得结果见表5。由表5可知,各组分的相对标准偏差(RSD)均小于2.0%,表明本法精密度较好。
用本法测量由铀矿石国家标准物质GBW 04101,GBW 04102和铀钼矿管理样M1、M2制成的4个玻璃样片,测定结果见表6。由表6可知,测量结果和标准值(或参考值)基本吻合。

表5 方法精密度试验结果 %
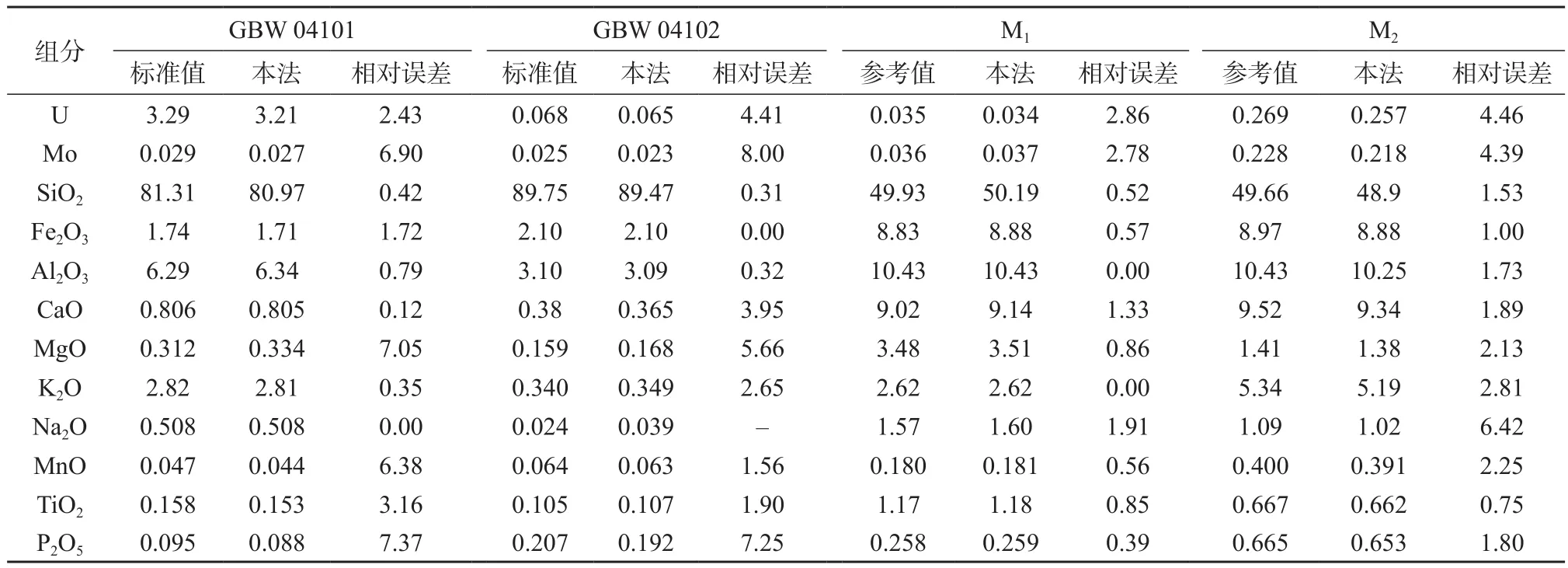
表6 方法准确度试验结果 %
3 结语
用熔融制样—X射线荧光光谱法同时测定铀矿中主次成分,在熔融制样过程中,预先在坩埚底部铺一层固体NH4NO3,可缩短预氧化时间,且更有利于保护坩埚不被腐蚀。该方法减化了化学法的复杂流程,降低了分析误差;同时,实验表明只要在标准物质充足的条件下,也可对铀钼矿中其它成分的测定进行探讨。
[1] 邢会敏,刘艳,陈刚,等.国外铀钼伴生矿综合回收技术[J].铀矿冶,2006,25(4): 186-191.
[2] 王洪明,孟晋,康绍辉,等.铀钼伴生矿的强化堆浸研究[J].铀矿冶,2003,31(3): 122-125.
[3] 刘国生,王速,王家和,等.毛洋头铀矿床淋浸液萃取分离铀钼工艺技术进展[J].铀矿冶,2000,19(3): 205-209.
[4] 郑英,樊保团,刘建,等.铀钼共生矿的细菌浸出试验研究[J].湿法冶金,2007,26(6): 75-79.
[5] 黄汉嘉,李占春.离子交换技术在火山热液铀钼类型矿石堆浸生产中的应用[J].铀矿地质,2007,23(3): 187-192.
[6] 梁冠杰.铀钼分离选冶试验研究[J].现代矿业,2010(3): 40-43.
[7] 成宝海,肖超,肖连生.铀钼分离远离与工艺[J].湿法冶金,2011,30(2): 99-102.
[8] EJ 267.2-1984 铀矿石中铀的测定三氯化钛还原钒酸铵氧化滴定法[S].
[9] EJ 297-1987 花岗岩、花岗岩铀矿石组份分析方法[S].
[10] GB/T 14352.2-2010 钨矿石、钼矿石化学分析方法 第2部分:钼量测定[S].
[11] 黄光明,薛蒙伟,韩鹏飞,等.密闭消解ICP-AES测定钨矿石和钼矿石中的9种组分[J].光谱实验室,2013,30(2): 956-962.
[12] 杨小丽,李小丹,杨梅. X射线荧光光谱法测定以钨和钼为主的多金属矿中主次成分[J].冶金分析,2013,33(8): 38-42.
[13] 田文辉,王中岐,张敏.能量色散X射线荧光光谱法测定钼矿石中钼铅铁铜[J].岩矿测试,2008,27(3): 235-236.
[14] 李红叶,许海蛾,李小莉,等.熔融制片-X射线荧光光谱法测定磷矿石中主次量组分[J].岩矿测试,2009,28 (4): 379-381.
[15] 吉昂,陶广义,卓尚军,等. X射线荧光光谱分析[M].北京:科学出版社2003: 164-165.
[16] 张建波,林力,王谦,等. X-射线荧光光谱法同时测定镍红土矿中主次成分[J].冶金分析,2008,28(1): 15-19.
[17] DZ/T 0130-2006 地质矿产实验室测试质量管理规范[S].
Simultaneous Determination of Major and Minor Components in Uranium-molybdenum Ores by X-ray Fluorescence Spectrometry with Fusion Sample Preparation
Liu Jinliang, Zhang Xin, Song Maosheng
(Research Institute No. 230, CNNC, Changsha 410007, China)
A method for simultaneous determination of U, Mo, SiO2, Fe2O3, Al2O3etc. in uranium-molybdenum ores by X-ray fluorescence spectrometry using fusion sample preparation with mix flux was developed. Li2B4O7-LiBO2was used as mixed flux, and NH4NO3was used as oxidizing agent, the mass ratio of samples with mixed flux was 1∶10. Except adding some uranium ore national standard substances, working curve was made by selecting artificial mixture of calibration samples and calendar year laboratory comparison of uranium molybdenum calibration sample. Matrix effects were corrected by the method of theoretical coefficients and the overlapping spectral line interference of the corresponding elements was deducted by Br Kα, Zr Kα, U Lα, Ba Lα, Zn Kαspectral lines. The precision of each component was less than 2.0% (n=10), the method with the relative error of 0.00-8.00% was applied to the determination of these components in national standard material and the results were in agreement with certified values. This method can meet the requirements of accurate and rapid measurement of uranium molybdenum ore in the production experiment.
X-ray fluorescence spectrometry; fusion sample preparation; uranium-molybdenum ores; major and minor components
O657.34
A
1008-6145(2016)05-0060-04
10.3969/j.issn.1008-6145.2016.05.016
*中国地质调查局全国铀资源调查评价工作项目(1212011220782)
联系人:柳金良;E-mail: 616160935@qq.com
2016-07-12
猜你喜欢
有色金属材料与工程(2022年4期)2022-10-27 11:43:22
粉末冶金技术(2021年3期)2021-07-28 06:26:42
矿产勘查(2020年4期)2020-01-05 02:34:33
震灾防御技术(2019年3期)2019-06-02 08:25:30
炎黄地理(2017年10期)2018-01-31 02:15:19
凿岩机械气动工具(2017年1期)2017-05-17 06:19:48
中国塑料(2016年10期)2016-06-27 06:35:22
中成药(2016年8期)2016-05-17 06:08:22
铜业工程(2015年4期)2015-12-29 02:48:44
中国钼业(2014年3期)2014-01-30 19:55:22
