
Exploring Genome-wide DNA Methylation Profiles Altered in Kashin-Beck Disease Using Infinium Human Methylation 450 Bead Chips*
2016-10-24 10:32SHIXiaoWeiSHIBoHuiLYUAiLiZHANGFengZHOUTianTianandGUOXiong
SHI Xiao Wei, SHI Bo Hui, LYU Ai Li, ZHANG Feng, ZHOU Tian Tian, and GUO Xiong,#
Exploring Genome-wide DNA Methylation Profiles Altered in Kashin-Beck Disease Using Infinium Human Methylation 450 Bead Chips*
SHI Xiao Wei1, SHI Bo Hui3, LYU Ai Li2, ZHANG Feng2, ZHOU Tian Tian2, and GUO Xiong2,#
To understand how differentially methylated genes (DMGs) might affect the pathogenesis of Kashin-Beck disease (KBD). Genome-wide methylation profiling of whole blood from 12 matched KBD and controls pairs was performed using a high-resolution Infinium 450 K methylation array. In total, 97 CpG sites were differentially methylated in KBD compared to the normal controls; of these sites, 36 sites were significantly hypermethylated (covering 22 genes) and 61 sites were significantly hypomethylated (covering 34 genes). Of these genes, 14 significant pathways were identified, the most significant P value pathway was type I diabetes mellitus pathway and pathways associated with autoimmune diseases and inflammatory diseases were included in this study. Subsequently, 4 CpG sites in HLA-DRB1 were validated using bisulfite sequencing polymerase chain reaction (BSP) in articular cartilage, and the results showed significant differences in the methylation status between KBD and controls,consistent with the results of the high-resolution array. These results suggested that differences in genome-wide DNA methylation exist between KBD and the controls, and the biological pathways support the autoimmune disease and inflammatory disease hypothesis of KBD.
Kashin-Beck disease (KBD) is an endemic and chronic osteochondropathy that is primarily distributed within a limited area that extends from southeastern Siberia to northeastern and southwestern China. Biogeochemical risk factors such as endemic selenium deficiency, high humic acid levels in drinking water, and the contamination of cereals with mycotoxin- producing fungi have long been known to cause KBD[1]. However, the etiology of KBD remains unclear.
Recent results from epidemiological and genetic studies suggest that interactions between environmental factors and susceptibility genes might play a role in the disease[2], and researchers have identified a set of abnormally expressed genes,proteins and pathways that are associated with KBD[3]. Differences in the phenotypes and the expression of genes between KBD patients and normal controls reflect a profound change that are in the processes involved in gene regulation at the transcriptional and post-transcriptional levels[3]. Epigenetic alterations comprise modifications of DNA and histones, which regulate gene expression through changes in chromatin structure[4]. Environmental factors are now known to directly or indirectly induce epigenetic changes, which modulate gene expression and are thus associated with changes in cell function[5]. As noted above, both genetic and environmental factors contribute to the etiology of KBD. However, to date, little is known regarding the role of epigenomics, particularly DNA methylation profiles, in the pathophysiology of KBD.
The Infinium Human Methylation 450 (450 K)BeadChip array includes 485,764 CpG sites from 21,231 genes, covering 96% of the CpG islands and 99% of the RefSeq genes, with multiple probes per gene from the University of California Santa Cruz (UCSC) database[6]. The application of this array indicates that the Infinium Human Methylation 450 BeadChip is an efficient, robust and affordable tool for assessing epigenetic changes across the human genome. In the current study, for the first time, we applied these arrays, to determine aberrant DNA methylation patterns of KBD in the Chinese Han population.
Twelve matched pairs of KBD and controls were included to explore alterations in genome-wide DNA methylation profiles. Radiographs of the right hand were taken for both KBD patients and healthy controls and read by veteran orthopedists. KBD was diagnosed according to the Chinese national diagnostic criteria for KBD (WS/T 207-2010). The healthy controls were defined as having neither KBD nor arthritis and were selected from the same KBD-endemic area. Fresh blood (5 mL) was collected from the antecubital vein in a fasting state. Total DNA was extracted using a Blood DNA Extraction Kit (Bei-Mei Co., Shaanxi, China). The study was performed in accordance with the Declaration of Helsinki and was approved by the Human Ethics Committee of Xi'an Jiaotong University, China. Informed written consent was obtained from each subject.
Bisulfite modification of 1.25 μg of DNA was conducted using an EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA) according to the manufacturer's procedure. The Infinium Methylation 450K assay was performed according to Illumina's standard protocol. The DNA methylation profiling data were processed using the Methylation Module of the GenomeStudio V1.9 software with the default parameters (Illumina). Before statistical analysis, the following pre-processing was applied: cell type composition was estimated using Houseman algorithm, remove probes with a detection P>0.05;adjust for age and gender balance; Color-bias adjustment and quantile normalization were performed on signal intensities as implemented in lumi; CpG sites on the sex chromosomes were removed to avoid gender specific methylation bias. Subsequently, an empirical Bayes-moderated t-test with Bonferroni correction for multiple testing was used to identify CpG sites that are differentially methylated between KBD and controls. A significant difference was defined as sites with a Bonferroni-corrected P value ≤0.05 which corresponds to a raw P value of ≤1.08×10-7. To investigate the biological relevance of these genes, a GO analysis was conducted using the FatiGO tool. Additionally, these genes were mapped to KEGG pathways and functional categories via Wikipathway analysis.
Additional, knee articular cartilage was obtained from 21 cases who had undergone amputation because of traffic accidents (Controls, 11 men and 10 women with a mean±SD age of 48.23±7.65 years)and from 22 KBD patients who underwent knee debridement or arthroplasty at a hospital (KBD, 10 men and 12 women with a mean±SD age of 51.00±8.30 years). Genomic DNA was extracted using an AllPrep DNA Mini kit (Qiagen). HLA-DRB1 has been reported to be associated with KBD[7], and the most significant P value CpG site was in HLA-DRB1; thus, 2 hypermethylated (cg15568074,16514085) and 2 hypomethylated (cg04026937,cg18052547) significant CpG sites in HLA-DRB1 were validated used bisulfite sequencing polymerase chain reaction (BSP). BSP was performed with 500 ng of genomic DNA from each sample using an EZ DNA Methylation-Gold kit (Zymo Research Corporation)according to the manufacturer's instructions.
The combined data from the 24 samples revealed that 97 CpG sites associated with 54 different genes had significant differences in DNA methylation between the KBD cases and the healthy controls. Among the significant CpG sites, 36 (37.1%)were significantly hypermethylated (covering 22 genes), and 61 (62.9%) were significantly hypomethylated (covering 34 genes). Most of the significant CpG sites, 50.0% (18/36) hypermethylated and 44.3% (27/61) hypomethylated CpG sites, are found in open sea (>6 kb from CpG islands) (Table 1). Based on the significant CpG sites, the fragments were further counted in which the significant CpG sites were all hypermethylation or all hypomethylation, in regions of promote, twelve genes were included in the DMRs (differentially methylated regions), including one miRNA. These results suggested that differences in genome-wide DNA methylation exist between KBD and the controls.
A functional analysis of the genes associated with the differentially methylated sites revealed a strong enrichment in the categories of ‘Biological processes' and ‘Molecular functions' (Figure 1). GO analysis revealed that the KBD-associated genes were highly involved in immune functions, and a number of the histocompatibility leukocyte antigen (HLA) complex genes were found to be associated with KBD. Of the methylated gene sites, the most significant P value was for HLA-DRB1. HLA-DRB1 has been reported to be associated with osteoarthritis (OA)[8], rheumatoid arthritis (RA)[9], and KBD[7]. The DMGs (differentially methylated genes) of HLA-DQA1,HLA-DRB5, HLA-DPB1, and HLA-DQB1 were all reported to be associated with RA. Our findings are consistent with those of previous studies, which support the association of HLA-DRB1 with KBD and the autoimmune disease hypothesis of KBD[7].
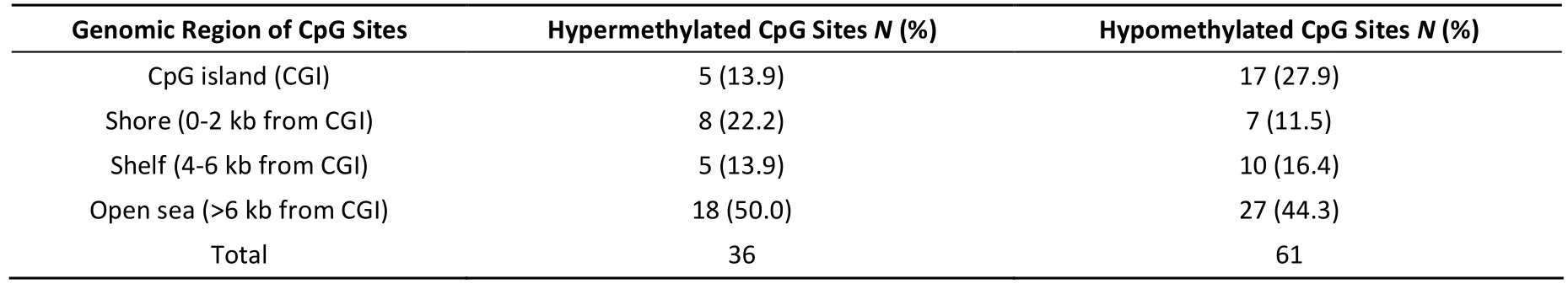
Table 1. Distribution of Genomic Regions for All Significantly Differentially Methylated CpG Sites in KBD Blood Cells Compared with Healthy Blood Cells

Table 2. KEGG Pathway Analysis of the Differentially Methylated and Expressed Genes
To further investigate the key pathways linked to these distinct genes, significant pathways associated with KBD were constructed according to the KEGG database. Our analysis revealed that these DMGs are widely involved in various cellular pathways, and 14 signaling pathways were detected and considered significant (q<0.05). These biological pathways are listed in Table 2. The top-ranked pathway is the KEGG ‘Type I DM' pathway (q=4.12×10-8). A previous meta-analysis identified a relationship between RA and the occurrence of DM[10]. KBD has been found to have overlapping phenotypes and pathologic changes similar to those of RA, and we speculate that the type I DM pathway may play a direct or indirect role in KBD, although few reports have linked this pathway to KBD. Both pathways associated with immune diseases (eg,allograft rejection, graft-versus-host disease,autoimmune thyroid disease, et al.) and those associated with inflammatory diseases (eg, viral myocarditis, asthma, staphylococcus aureus infection, et al.) were significantly enriched in the differentially methylated profiles. These results suggest that immune and inflammatory processes may be key regulators of KBD, although further,well-designed studies with a larger sample sizes are required to confirm this idea.
We verified 4 CpG sites in HLA-DRB1 by BGS in articular cartilage. Of the 2 hypermethylated CpG sites, the methylation levels detected in KBD and controls were 72.7% (16/22) vs. 23.8% (5/21) and 72.7% (16/22) vs. 14.3% (3/21), (X2=10.28, 14.88,both P<0.05). For the 2 hypomethylated CpG sites, the methylation levels detected in KBD and controls were 36.4% (8/22) vs. 71.4% (15/21); 22.7% (5/22) vs. 61.9% (13/21), (χ2=15.31, 6.76; both P<0.05). Abnormal methylation of HLA-DRB1 is found in KBD,which was consistent with the previous results of altered DNA methylation profiles. These results suggest that our data are reliable, despite certain limitations in this study. The sample populations were small, the pathway analyses were based on a limited number of genes, and the positive results were confirmed only by BSP of HLA-DRB1, although other genes and more detailed mechanistic studies are still worthy of further investigation.
In summary, we have comprehensively characterized the genome-wide DNA methylation patterns occurring in KBD and identified a set of CpG sites, genes and pathways correlated with KBD. Our findings support the autoimmune disease and inflammatory disease hypothesis of KBD. We believe that the robust data presented here provide valuable information for better understanding the molecular mechanisms involved in the multistep development of KBD.
For the sample collection, the authors acknowledge help from the Centers for Disease Control and Prevention of Linyou counties.
#Correspondence should be addressed to GUO Xiong,Tel: 86-29-82655091, E-mail: guox@mail.xjtu.edu.cn
Biographical note of the first author: SHI Xiao Wei,female, born in 1971, PhD, majoring in molecular mechanisms of cartilage and bone.
1. Suete C, Moreno R, Chassear C, et al. Epidemiological support for a multifactorial aetiology of Kashin-Beck disease in Tibet. Int Orthop, 2001; 25, 180-7.
2. Zhang F, Guo X, Wang WZh, et al. Genome-wide gene expression analysis suggests an important role of hypoxia in the pathogenesis of endemic osteochondropathy Kashin-Beck Disease. PLos One, 2011; 6, e22983.
3. Guo X, Ma WJ, Zhang F, et al. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthritis Cartilage, 2014; 22, 1774-83.
4. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell, 2007; 128, 669 -81.
5. De la Rica L, Urquiza JM, Gómez-Cabrero D, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun, 2013; 41, 6-16.
6. Dedeurwaerder S, Defrance M, Calonne, et al. Evaluation of the Infinium Methylation 450K technology. Epigenomics, 2011; 3,771-84.
7. Shi Y, Lu F, Liu X, et al. Genetic variants in the HLA-DRB1 gene are associated with Kashin-Beck disease in the Tibetan population. Arthritis Rheum, 2011; 63, 3408-16.
8. Sakkas LI, Platsoucas CD. The role of T cells in the pathogenesis of osteoarthritis. Arthritis Rheum, 2007; 56, 409-24.
9. Stark K, Straub RH, Blazicková S, et al. Genetics in neuroendocrine immunology: implications for rheumatoid arthritis and osteoarthritis. Ann NY Acad Sci, 2010; 1193, 10-4.
10.Jiang P, Li H, Li X. Diabetes mellitus risk factors in rheumatoid arthritis: a systematic review and meta-analysis. Clin Exp Rheumatol, 2015; 33, 115-21.
10.3967/bes2016.072
December 10, 2015;
*This research was supported by grants from the National Natural Science Foundation of China (No. 81273007).
1. Department of Pediatrics, The First Affiliated Hospital of the Medical College of Xi'an Jiaotong University, Xi'an 710061, Shannxi, China; 2. School of Public Health, Health Science Center, Xi'an Jiaotong University, Key Laboratory of Environment and Gene Related Diseases of Ministry of Education, Key Laboratory of Trace Elements and Endemic Diseases of Ministry of Health, Xi'an 710061, Shannxi, China; 3. Department of Oncosurgery, The First Affiliated Hospital of the Medical College of Xi'an Jiaotong University, Xi'an 710061, Shannxi, China
Accepted: May 5, 2016
 Biomedical and Environmental Sciences2016年7期
Biomedical and Environmental Sciences2016年7期
- Biomedical and Environmental Sciences的其它文章
- ldeal Cardiovascular Health Metrics and Coronary Artery Calcification in Northern Chinese Population:A Cross-sectional Study*
- p21 is Responsible for Ionizing Radiation-induced Bypass of Mitosis*
- Autophagy Attenuates MnCl2-induced Apoptosis in Human Bronchial Epithelial Cells*
- Lack of Association between rs4331426 Polymorphism in the Chr18q11.2 Locus and Pulmonary Tuberculosis in an Iranian Population*
- Detection of Multi-drug Resistant Acinetobacter Lwoffii Isolated from Soil of Mink Farm*
- Green Tea Polyphenols Alleviate Autophagy Inhibition Induced by High Glucose in Endothelial Cells*