
HPLC法快速测定竹凉席中的罗丹明B
2016-09-28 03:01卢桂英程水连何建国宋艳春
中国新技术新产品 2016年16期
卢桂英 程水连 何建国 宋艳春
(湖南省益阳市产商品质量监督检验所,湖南 益阳 413000)
HPLC法快速测定竹凉席中的罗丹明B
卢桂英 程水连 何建国 宋艳春
(湖南省益阳市产商品质量监督检验所,湖南 益阳 413000)
建立了快速测定竹凉席中染色剂罗丹明B的含量的方法。以甲醇水溶液超声提取,用高效液相色谱法(荧光检测器)测定,峰面积外标法定量,在优化条件下,罗丹明B的质量浓度在0.5~100.0ng/mL范围内与色谱峰面积呈现良好的线性关系,线性相关系数r=0.9999,检出限为3.0ng/L,加标回收率为87.0%~100.2%,得到罗丹明B的质量浓度和保留时间的相对标准偏差分别为1.24%和0.05%。该方法样品处理简单、快速,适用于竹凉席中罗丹明B的日常检测。
罗丹明B;高效液相色谱法;荧光检测器;竹凉席
罗丹明B,又称玫瑰红B,或碱性玫瑰精,俗称花粉红,是一种具有鲜桃红色的人工合成染料。罗丹明B在溶液中有强烈的荧光,用作实验室中细胞荧光染色剂及有色玻璃、特色烟花爆竹等行业。本研究建立了一种准确、灵敏的检测竹凉席中的罗丹明B含量的方法,该方法样品前处理简单、罗丹明B的色谱峰形好、定量结果具有很好的重现性、精密度,该方法有着重要的实际应用价值。
1.试验部分
1.1仪器和试剂
WH-400超声清洗器(济宁万和超声电子设备有限公司);LC-MS2020液相色谱仪,带荧光检测器,(日本岛津公司);LP502A型分析天平,感量0.0001g,(常熟市百灵天平仪器有限公司);中草药粉碎机(天津市泰斯特仪器有限公司);标准筛100目;0.45μm有机微孔滤膜;超纯水器(艾柯公司)。
1.2方法
1.2.1标准曲线各浓度溶液的配制
标准溶液的配置:精密称取罗丹明B0.0500g置于100mL棕色容量瓶中,用甲醇溶解并准确定容至刻度,摇匀,配成质量浓度为0.5mg/mL的罗丹明B标准储备液。标准工作液的配制:用流动相将上述标准液稀释为0.5ng/mL,1.0ng/mL,5.0ng/mL,10ng/mL,50ng/mL,100ng/mL,的工作液。此工作液现配现用。
1.2.2样品的制备
取烘干至恒重的竹凉席块切小,放入中草药粉碎机中粉碎,过60目的分子筛,取混合均匀的样品2g,放入100mL的容量瓶中,加色谱纯甲醇至刻度,超声提取30min,静置5min后,取上清液过0.45μm有机微孔滤膜后,用液相色谱仪测定。
1.2.3色谱分析条件
色谱柱:C18,柱长150mm,内径4.6mm,粒度5μm;柱温:35℃。
进样量:10μL;流速:1.0mL/min;流动相:甲醇和水(比例为60:40)。
荧光条件:激发波长(Ex)550nm,发射波长(Em)580nm。
2.结果与讨论
2.1检测器及检测波长的选择
因罗丹明B的甲醇溶液有强烈的荧光,相比紫外检测器,荧光检测器的灵敏度更高,而荧光检测器相比于质谱检测器,价格更优惠,实验室使用更普及些,因此选用荧光检测器作为高效液相的检测器测定罗丹明B;通过对罗丹明B的标准溶液进行光谱扫描可知,罗丹明B在550nm处吸收峰最大,因此将选定为550nm为激发波长(Ex),580nm为发射波长(Em)。
2.2样品提取条件的选择
罗丹明B是酸碱两性化合物,易溶于水、甲醇、乙腈等。其中甲醇溶液呈现玫瑰红色,有强烈的荧光,故选用纯的甲醇提出。因罗丹明B是易溶的物质,而且超声提取是最简单快捷的提取方法,所需要时间短,溶剂量少而且提取完全,因此采用超声提取法。超声提取后,样品溶液直接进液相色谱分析,待测组分的损失少,回收率也很高。

表1 精密度试验结果
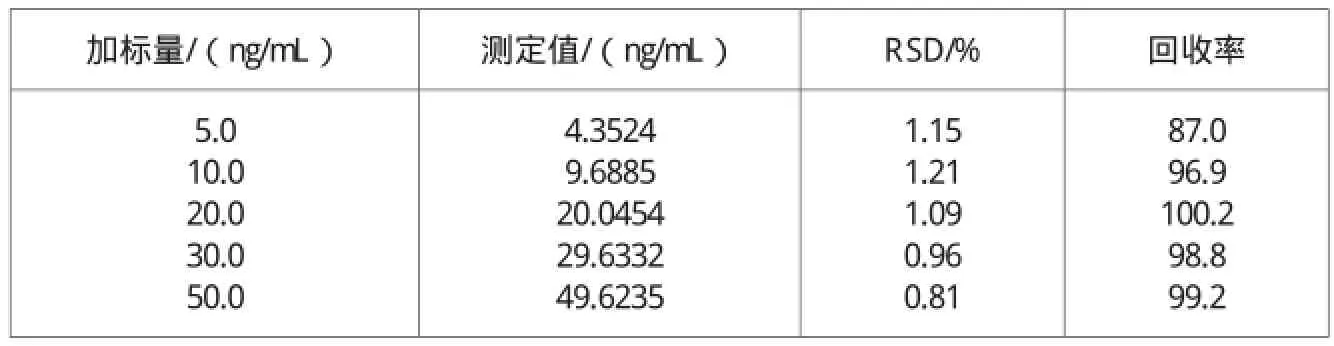
表2 回收试验结果(n=6)
2.3色谱条件的选择
根据罗丹明B的性质采用反相液相色谱法,C18,(150mm×4.6mm,5μm)色谱柱进行分离,该色谱柱由于粒径小,比表面积大,柱效显著提高,色谱分析比较理想。流动相选择:分别尝试了不同比例的甲醇-水溶液进行洗脱,并尝试了梯度洗脱方式,发现当甲醇与水的比例为60∶40,进行等度洗脱时,峰形很好,如图1和图2所示,图1为罗丹明B标准色谱图,出峰时间为13.157min。图2为样品的色谱图,出峰时间为13.170min。因此选择了甲醇-水为60∶40的体系作为流动相,等度洗脱方式。
2.4线性方程及检出限
用罗丹明B标准储备溶进行稀释,配置以下浓度的罗丹明B工作液:0.5ng/mL,1.0ng/mL,5.0ng/mL,10ng/mL,50ng/mL,100ng/mL,6个水平的工作液。对每个工作浓度液分别平行测定3次,以罗丹明B标准溶液的质量浓度为横坐标(X),以各个浓度水平的色谱峰峰面积为纵坐标(Y)进行线性拟合。结果表明,罗丹明B标准溶液的质量浓度在0.5~100ng/mL范围内与相应色谱峰峰面积呈现良好的线性关系,线性方程为Y=47540X+6123(r=0.9999)。
同等色谱条件下进行空白试验,对空白样平行测定10次,计算相应的标准偏差,以空白的标准偏差的3倍值除以线性方程的斜率,得方法的检出限为3.0ng/g.
2.5精密度试验
对20.0ng/mL的罗丹明B标准溶液进行8次平行测定,测定结果列于表1.并计算8次结果的相对标准偏差,得到罗丹明B的质量浓度和保留时间的相对标准偏差分别为1.24%和0.05%,表明该方法精密度良好。
2.6回收试验
采用没有用罗丹明B染色的竹凉席中添加罗丹明B标准溶液的方法进行回收试验。准确称取相同的空白凉席试样5份,分别加入5.0ng/mL,10.0ng/mL,20.0ng/mL, 30.0ng/mL,50.0ng/mL5个不同水平的罗丹明B标准溶液,进行与样品相同的提取处理和测定,回收试验结果见表2。由表2可知,在添加质量浓度为5.0~50.0ng/mL时罗丹明B的回收率为87.0%~100.2%,相对标准偏差在0.81%~1.21%之间。
结语
研究通过高效液相色谱法测定染色竹凉席中的罗丹明B含量的结果表明,该方法:前处理简单快速、灵敏度高、准确性好、可操作性强,能满足于竹凉席中非法添加染色剂罗丹明B冒充碳化竹凉席的日常检测。
[1]陈国珍,黄贤智,郑朱梓,等.荧光分析法(第二版)[M].北京:北京科学出版社,1990:69.
[2]卢士英,邹明强.食品中常见的非食用色素的危害与检测[J].中国仪器仪表,2009(8):45-50.
O657
A
猜你喜欢
科学技术创新(2021年19期)2021-07-16
海峡姐妹(2020年8期)2020-08-25
幸福(2019年21期)2019-08-20
科技创新与应用(2017年1期)2017-05-11
科技与创新(2016年10期)2016-05-28
化工进展(2015年3期)2015-11-11
湖北理工学院学报(2015年1期)2015-02-27
测绘通报(2014年3期)2014-08-16
山西大同大学学报(自然科学版)(2014年2期)2014-01-23
化学分析计量(2013年5期)2013-03-11
