
CeO2-活性炭催化臭氧化降解水中草酸的研究
2016-09-15 06:13:06曾凡炎王淑静
工业水处理 2016年8期
杨 郭,曾凡炎,王淑静,马 燮,黄 斌
(1.四川理工学院材料与化学工程学院,四川自贡643000;2.北京大学定量生物学中心,北京100871)
CeO2-活性炭催化臭氧化降解水中草酸的研究
杨郭1,曾凡炎1,王淑静2,马燮1,黄斌1
(1.四川理工学院材料与化学工程学院,四川自贡643000;2.北京大学定量生物学中心,北京100871)
利用CeO2的储氧和释氧能力,以高活性的活性炭作为催化剂载体,采用等体积浸渍法制备CeO2-活性炭催化剂。分别用氮气低温吸附/脱附、XRD、XPS对CeO2-活性炭催化剂进行表征。以CeO2-活性炭作为催化剂,草酸作为模型化合物,催化臭氧化降解草酸的去除率达到77%。Ce组分的加入能显著提高草酸的去除率。Ce不仅能够提高反应体系中羟基自由基的浓度,还有利于O3分子直接氧化草酸。CeO2-活性炭催化剂经过3次重复使用后,草酸去除率保持在70%以上,具有较好的稳定性。
草酸;活性炭;二氧化铈;臭氧化;催化降解
草酸是一种重要的化工原料,广泛应用于医药、化工等领域〔1〕。草酸也是多种有机物污染物经臭氧催化氧化处理后的难降解中间产物,和有机污染物的矿化程度直接相关〔2〕。因此,臭氧催化氧化反应中草酸去除率的高低与催化剂活性密切相关。
臭氧催化氧化反应是利用催化剂提高臭氧的利用率,促进O3产生更多的羟基自由基,加快对有机物的分解速度。研究表明,活性炭(AC)能加速O3的分解产生·OH。U.Jans等〔3〕向含O3的溶液中添加微量活性炭或炭黑能明显加快O3分解生成·OH的速率,12 min内O3的分解率提高到88.9%。F.J.Beltrán等〔4〕对活性炭催化分解臭氧动力学的研究也证实,活性炭可加快O3在水溶液中的分解速率。此外,金属氧化物也被证实能提高O3对有机污染物的降解能力,如 TiO2〔5〕、Fe2O3〔6〕、MnO2〔7〕、Co3O4〔8〕等均能提高O3对草酸的去除率。
近年来,含CeO2材料逐渐应用到很多领域,如发光材料、光催化、紫外吸收材料、汽车尾气净化催化剂等〔9〕,但将CeO2应用于水中草酸的研究还鲜有报道。相对于已经报道的金属氧化物,CeO2是一种N型半导体,具有较强的氧化还原能力,Ce很容易在Ce3+和Ce4+间转换,具有较高的储氧和释氧能力,当CeO2经过高温处理后,易造成氧缺位,形成潜在的活性中心。
笔者采用活性炭作为载体,以硝酸铈作为前驱体,采用等体积浸渍法制备了CeO2-活性炭(CeO2-AC)催化剂。利用AC和铈氧化物的协同作用,提高对草酸的去除率,并对CeO2-AC催化臭氧化降解草酸的机理进行了研究。
1 试验部分
1.1试剂与仪器
活性炭购于山西华瑞活性炭有限公司;Ce(NO3)3·6H2O、草酸等试剂均为分析纯。
SSA-4200比表面积测定仪,彼奥德电子技术有限公司;KTL型管式炉,南京南大仪器有限公司;NS-110X30水浴恒温摇床,上海世平实验设备有限公司;pHS-25型酸度计,成都方舟科技开发公司;FA2004N型电子天平,上海民桥精密科学仪器有限公司;UV-2000型紫外可见分光光度计,尤尼柯仪器有限公司;台式COD测定仪,北京双晖京承电子产品有限公司;ZCS臭氧发生器,秦皇岛展坤消毒设备有限公司;Agilent 1100高效液相色谱仪,安捷伦科技有限公司。
1.2催化剂的制备
活性组分铈前躯体为Ce(NO3)3·6H2O,在不断搅拌下逐滴加入到适量的活性炭(0.15~0.074 mm,100~200目)中,采用等体积浸渍法浸渍过夜,100℃干燥后,于管式炉中在300℃下煅烧2 h后降至室温,整个煅烧过程均在氮气保护下进行,氮气流量为60 mL/min。
1.3催化剂的表征
1.3.1比表面积和孔径分布
活性炭载体和催化剂样品的孔结构测试在NOVA 2200e型氮气吸附仪上进行,样品的比表面积通过BET方程计算,总的孔体积通过氮气在相对压力为0.99时的吸附量转换为相应体积获得;中孔体积、中孔的比表面积和孔径分布通过BJH方法测得。
1.3.2粉末X射线衍射(XRD)
XRD测试由X’pert pro MPO X射线衍射仪(荷兰飞利浦公司)进行。测试条件:Cu靶(Kα),扫描角度:5~70°。加速电压40 kV,管电流35 mA,扫描速度为0.15(°)/s,扫描步幅为0.3°。
1.3.3X射线光电子能谱(XPS)
XPS表征在KratosXSAM800型X射线光电子能谱仪上进行,采用AlKαX射线作为激发源,光子能量为1486.6eV;靶功率为250 W,电压12.5 kV;化学位移的校正采用内标法,以有机污染碳的C1s的标准结合能284.6 eV为基准进行校准。
1.4草酸的催化臭氧化试验
草酸催化臭氧化试验在2 L玻璃反应釜中进行,草酸溶液体积1.5 L,初始质量浓度为500 mg/L,催化剂用量为250 mg/L。氧气经过臭氧发生器产生O3,氧气流量为200 mL/min,O3进入反应器的质量浓度为40 mg/L,O3经过玻璃反应器底部的气体分布装置与溶液鼓泡接触。溶液中草酸的测定采用高效液相色谱进行分析,色谱柱为C18XDB(5 μm,4.6 mm×150 mm);DAD检测器(波长254 nm);V(乙腈)∶V〔磷酸溶液(pH=3)〕为10∶90。草酸的剩余率按照式(1)计算。

式中:X——草酸的剩余率,%;
C——草酸任意时刻的质量浓度,mg/L;
C0——草酸的初始质量浓度,mg/L。
2 结果与讨论
2.1催化剂的表征
2.1.1活性炭和催化剂的孔结构
活性炭(AC)和CeO2-AC催化剂的比表面积和孔体积参数见表1。
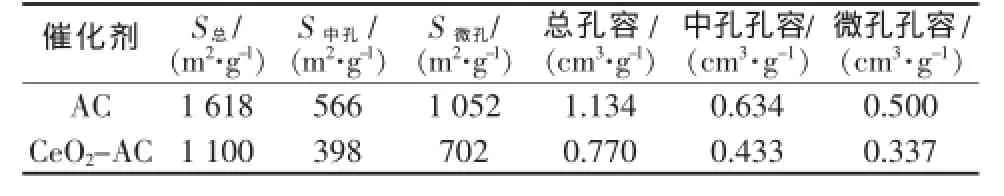
表1 AC和CeO2-AC催化剂的孔结构参数
由表1可见,催化剂CeO2-AC的比表面积为1 100 m2/g,孔体积为0.770 cm3/g。和活性炭相比,中孔和微孔的比表面积分别下降了30%和33%。考虑到煅烧温度较低,对活性炭的孔结构影响较小,因此CeO2-AC的比表面积和孔体积下降主要是CeO2颗粒同时进入了活性炭的微孔和中孔造成的。活性炭载体和催化剂CeO2-AC的孔径分布如图1所示。

图1 活性炭和CeO2-AC的孔径分布
活性炭表面负载Ce后,在孔径4 nm处的峰高和峰面积明显下降,这表明CeO2-AC催化剂的比表面积和孔体积比活性炭载体小。
2.1.2粉末X射线衍射(XRD)
有文献报道活性炭负载CeO2催化剂的特征衍射峰分别位于28.5°、33.1°、47.5°、56.3°、59.0°〔10〕。催化剂CeO2-AC在28.5°和47.5°有明显的衍射峰,但在33.1°、56.3°以及59.0°未出现明显的衍射峰。这可能是催化剂中Ce的负载量过低造成的〔11〕。当Ce的负载量低于10%,仅仅出现2个峰(28.5°、47.5°),当Ce的负载量超过10%后,28.5°、47.5°、56.3°均会出现特征衍射峰。为进一步确认Ce在催化剂载体表面的存在,采用XPS分析 Ce在活性炭载体表面的含量,同时表征Ce在活性炭表面的存在形式。
2.1.3催化剂X射线光电子能谱(XPS)
20世纪90年代,珠海市委、市政府确立了“一港带全局”的发展战略,珠海港作为国家定位的24个主要枢纽港之一,肩负着打造华南国际港口物流中心光荣使命,经过20多年的发展,一个亿吨大港已经在珠海西部强势崛起。在“一带一路”倡议和“发展粤港澳大湾区”战略背景下,珠海市迎来了前所未有的发展机遇,珠海具备区位、政策、环境、产业优势,正积极构建珠港澳港航物流生态圈,实现粤港澳大湾区港口群协同发展。
CeO2-AC催化剂的Ce XPS谱图如图2所示。
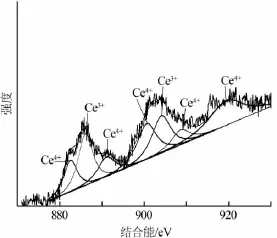
图2 CeO2-AC催化剂的Ce XPS谱图
根据文献〔12〕,Ce 3d XPS谱图可以拟合出7个峰,Ce3+的峰对应的结合能为886.28、904.02 eV,Ce4+对应的峰分别为883.60、889.71、889.25、901.04 eV,结合能在917eV处被认为是CeO2。活性炭表面Ce的存在形式有Ce3+和Ce4+,其中Ce3+的质量分数为36.3%,Ce4+占Ce总量的63.7%。这说明采用等体积浸渍后,经过300℃煅烧可以向活性炭的表面同时引入Ce3+和Ce4+,且催化剂表面主要以Ce4+的形式存在。
2.2草酸的催化臭氧化试验
草酸臭氧化、AC和CeO2-AC催化剂对草酸的吸附以及AC、CeO2-AC催化臭氧化降解草酸过程中,任意时刻的草酸剩余率随时间的变化曲线如图3所示。
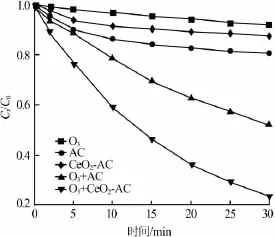
图3 吸附、臭氧化、催化臭氧化过程中草酸剩余率
从草酸的臭氧化降解曲线可以看出:反应30 min 后C/C0约为0.97,说明单独使用O3对草酸的降解能力十分有限。比较AC和CeO2-AC的吸附曲线,AC的吸附量稍高于 CeO2-AC,这主要是因为在CeO2-AC催化剂的制备过程中,CeO2颗粒进入到AC的微孔和中孔中,造成CeO2-AC的比表面积降低。因此CeO2-AC对草酸的吸附能力要低于AC。由图3可知,单独臭氧化及吸附过程并不能有效去除草酸。
分别以AC、CeO2-AC作为催化剂,采用催化臭氧化降解草酸时,30 min后草酸剩余率分别达到53%、27%,表明AC和CeO2-AC能显著提高O3对草酸的降解能力;并且相比AC,CeO2-AC作催化剂时对草酸的去除率提高了26%,可见Ce能提高对草酸的去除效果。
对S.Avramescu等〔13〕提出的臭氧催化降解草酸的动力学方程进行积分,可得:

kt——草酸氧化反应的总速率常数,min-1。
由式(2)可知草酸的催化臭氧化反应属于一级动力学,ln(CBt/CB0)与t呈线性关系,直线的斜率即为总速率常数。对单独臭氧化、AC和CeO2-AC催化臭氧化草酸的ln(CBt/CB0)~t关系作图,见图4。3个反应体系下的总速率常数(kt)和拟合的线性相关系数(R2)见表2。
由表2可见O3+CeO2-AC催化体系的总速率常数是O3+AC催化体系的2倍多,说明CeO2-AC催化剂具有较好的催化活性。

图4 单独臭氧化、AC和CeO2-AC催化臭氧化草酸的ln(CBt/CB0)~t
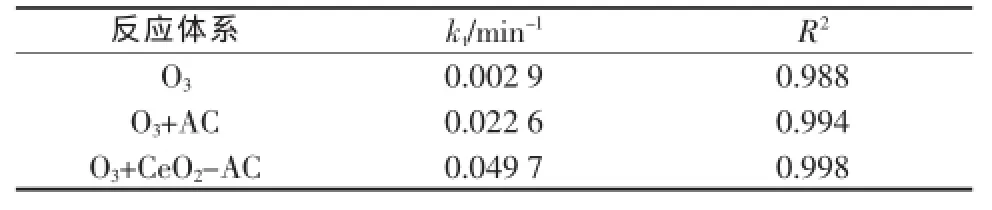
表2 草酸降解的一级动力学拟合结果
2.3叔丁醇对草酸去除率的影响
在催化臭氧化降解有机物的过程中,O3分子与草酸反应的同时,溶液中的自由基也会与草酸反应〔14〕。向反应溶液中加入叔丁醇能淬灭反应过程中产生的自由基〔15〕。考察了单独臭氧化、AC催化臭氧化及CeO2-AC催化臭氧化过程中叔丁醇对草酸浓度的影响,见图5。
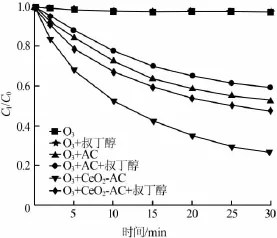
图5 叔丁醇对草酸去除效果的影响
如图5所示,对于单独臭氧化过程,叔丁醇的加入对草酸的降解基本没有影响。对于AC臭氧化过程,添加叔丁醇后,草酸剩余率明显增加,但仍然达到60%。因此,活性炭表面的活化O3分子与草酸之间的反应是草酸降解的主要途径〔16〕。对于CeO2-AC催化臭氧化过程,叔丁醇的加入造成草酸剩余率由27%增加到48%,表明草酸降解过程中有自由基参与,此外O3分子的直接氧化过程也具有重要作用。由此可见,Ce组分的添加不仅增加溶液中自由基浓度,还能够提高O3直接氧化草酸的能力。
2.4温度对草酸去除率的影响
不同温度下AC和CeO2-AC催化臭氧化降解草酸的效果如表3所示。

表3 反应温度对草酸去除效果的影响
由表3可见,反应温度的增加有利于草酸的去除,这主要是由于温度升高提高了反应动力学常数。此外,当温度从20℃升高到30℃,草酸的降低幅度要高于30℃增加到40℃的幅度,这是O3的溶解度随温度的升高而逐渐减小引起的。因此,过高的温度并不利于草酸的催化臭氧化反应。
2.5溶液pH对草酸去除率的影响
不同初始pH条件下AC和CeO2-AC催化臭氧化降解草酸的效果如图6所示。
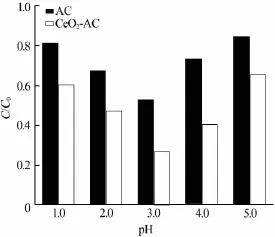
图6 溶液pH对草酸去除效果的影响
由图6可见,随着溶液pH的增加,AC和CeO2-AC降解草酸的能力均呈现先逐渐增加后减小的趋势。这与草酸分子在溶液中的存在形式以及催化剂表面电荷性质变化有关。草酸的两级解离常数分别为1.2、4.2。当溶液pH>1.2时,草酸发生一级电离,当溶液pH>4.2时,草酸完全解离成草酸根。
当溶液pH<3时,随着溶液pH的增加,更多的草酸发生一级电离,此时催化剂表面带正电荷,在静电引力作用下,大量草酸阴离子吸附在催化剂表面,造成草酸剩余率较小。当溶液pH继续增加,溶液中出现更多的草酸根离子,与此同时,由于pH的增加造成催化剂表面逐渐由正电荷变为负电荷,在静电斥力作用下,吸附在催化剂表面的草酸离子逐渐减少,造成草酸剩余率增加。此外,O3分子在较高pH溶液中更易分解也是造成草酸剩余率较高的一个原因。
2.6CeO2-AC催化剂的重复使用
在应用过程中,催化剂会发生活性组分溶出、催化剂表面活性位点破坏等问题,这会导致催化剂的活性显著降低。为考察CeO2-AC催化剂的重复使用性能,每次实验结束后过滤催化剂,重新加入到新鲜草酸溶液中进行重复试验。结果发现,催化剂重复使用3次后草酸剩余率稍有增加,但草酸剩余率均<30%,表明CeO2-AC催化剂的稳定性较好。
3 结论
(1)向AC表面引入Ce组分制备出的CeO2-AC催化剂在草酸催化臭氧化反应中表现出较高的活性,CeO2-AC对草酸的去除率约为77%,比AC反应体系高30%;CeO2-AC降解草酸的动力学速率常数是AC反应体系的2倍多。CeO2-AC催化剂的高活性主要是由于Ce在提高溶液中自由基浓度的同时,也促进O3与草酸分子直接发生氧化反应。
(2)温度升高有利于草酸剩余率的降低,但由于O3溶解度的原因,过高的温度不利于O3对草酸的降解。pH会影响草酸分子在溶液中的存在形式和催化剂表面电荷性质,当溶液pH为3时,CeO2-AC催化剂的活性最高。CeO2-AC催化剂重复使用3次后,草酸去除率仍高于70%,CeO2-AC催化剂具有较好的稳定性。
[1]王龙耀,王丽娟,王岚.改性活性白土负载Fe(Ⅱ)催化臭氧化去除草酸的研究[J].工业水处理,2014,34(3):43-46.
[2]Beltrán F J,Rivas F J,Montero-de-Espinosa R.A TiO2/Al2O3catalyst to improve the ozonation of oxalic acid in water[J].Applied Catalysis B:Environmental,2004,47(2):101-109.
[3]Jans U,Hoigné J.Activated carbon and carbon black catalyzed transformation of aqueous ozone into OH-radicals[J].Ozone Science& Engineering,1998,24(1):67-90.
[4]Beltrán F J,Rivas J,Álvarez P,et al.Kinetics of heterogeneous catalytic ozone decomposition in water on an activated carbon[J].Ozone Science&Engineering,2002,24(4):227-237.
[5]Beltrán F J,Rivas J,Montero-de-Espinosa R.Catalytic ozonation of oxalic acid in an aqueous TiO2slurry reactor[J].Applied Catalysis B:Environmental,2002,39(3):221-231.
[6]Jung H,Kim J W,Choi H,et al.Synthesis of nanosized biogenic magnetite and comparison of its catalytic activity in ozonation[J].Applied Catalysis B:Environmental,2008,83(3/4):208-213.
[7]Tong Shaoping,Liu Weiping,Leng Wenhua,et al.Characteristics of MnO2catalytic ozonation of sulfosalicylic acid and propionic acid in water[J].Chemosphere,2003,50(10):1359-1364.
[8]Álvarez P M,Beltrán F J,Pocostales J P,et al.Preparation and structural characterization of Co/Al2O3catalysts for the ozonation of pyruvic acid[J].Applied Catalysis B:Environmental,2007,72(3):322-330.
[9]Krishna K,Buenolopez A,Makkee M,et al.Potential rare earth modified CeO2catalysts for soot oxidation:I.Characterisation and catllytic activity with O2[J].Applied Catalysis B:Environmental,2007,75(3/4):189-200.
[10]罗小涛,李金林.活性炭负载钴纳米催化剂的制备与表征[J].化学与生物工程,2010,27(8):24-27.
[11]Chen Honglin,Zhang Xiaoming,Feng Yujun,et al.Efficient degradation of fulvic acids in water by catalytic ozonation with CeO2/ AC[J].Journal of Chemical Technology&Biotechnology,2014,89(2):322-327.
[12]Faria P C C,Orfào J J M,Pereira M F R.A novel ceria-activated carbon composite for the catalytic ozonation of carboxylic acids[J]. Catalysis Communications,2008,9(11/12):2121-2126.
[13]Avramescu S,Bradu C,Udrea I,et al.Degradation of oxalic acid from aqueous solutions by ozonation in presence of Ni/Al2O3catalysts[J].Catalysis Communications,2008,9(14):2386-2391.
[14]Staehelin J N M,Hoigne J.Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions[J].Environmental Science and Technology,1985,19(12):1206-1213.
[15]王列,姚玉元,孙利杰,等.活性炭纤维耦合柠檬酸铁在中性pH条件下活化双氧水降解染料[J].化学学报,2013,71(12):1633-1638.
[16]Faria P C C,Orfao J J M,Pereira M F R.Activated carbon catalytic
ozonation of oxamic and oxalic acids[J].Applied Catalysis B:Environmental,2008,79(3):237-243.
yangguo0813@163.com。
Research on the degradation of oxalic acid in water by CeO2-activated carbon catalytic ozonation
Yang Guo1,Zeng Fanyan1,Wang Shujing2,Ma Xie1,Huang Bin1
(1.College of Materials and Chemical Engineering,Sichuan University of Science and Engineering,Zigong 643000,China;2.Center for Quantitative Biology,Peking University,Beijing 100871,China)
Utilizing the oxygen storage and oxygen release capacities of CeO2,and using activated carbon with high activity as catalyst carriers,CeO2-activated carbon catalyst has been prepared by isometric impregnation method.N2low temperature adsorption/desorption,XRD and XPS are used respectively for its characterization.Using CeO2-activated carbon(CeO2-AC)as catalyst,oxalic acid as model compound,the removing rate of oxalic acid degraded by catalytic ozonation reaches 77%.Adding Ce component can improve the removing rate of oxalic acid obviously.Ce can not only improve the concentration of hydroxyl radicals in the reaction system,but also is advantageous to oxalic acid to be oxidized directly by O3molecules.After CeO2-AC has been used three times repeatedly,the removing rate of oxalic acid stays at above 70%,having better stability.
oxalic acid;activated carbon;cerium oxide;ozonation;catalytic degradation
X703
A
1005-829X(2016)08-0055-05
四川省教育厅科研基金(14ZB0209);四川理工学院培育项目(2013PY02)
杨郭(1980—),高级实验师,博士,硕士生导师。E-mail:
2016-07-13(修改稿)
猜你喜欢
发明与创新(2023年30期)2023-10-11 01:37:12
小学生学习指导(高年级)(2023年3期)2023-03-31 06:03:22
小学生学习指导(高年级)(2022年3期)2022-03-29 07:49:16
童话世界(2020年32期)2020-12-25 02:59:18
中学生数理化·八年级物理人教版(2019年3期)2019-04-25 06:21:00
小学生导刊(高年级)(2017年2期)2017-06-10 02:40:42
材料科学与工程学报(2016年4期)2017-01-15 13:35:34
饮食科学(2016年3期)2016-07-04 15:12:40
饮食科学(2016年3期)2016-07-04 15:12:27
应用化工(2014年1期)2014-08-16 13:34:08
