
PCR扩增循环数对细菌群落多样性测序分析的影响
2016-09-13 08:37安云鹤高丽娟李俊博田彦捷王金龙郑学娟武会娟
生物工程学报 2016年8期
安云鹤,高丽娟,李俊博,田彦捷,王金龙,郑学娟,武会娟
1 北京市理化分析测试中心,北京 1000892 北京市基因测序与功能分析工程技术研究中心,北京 100094
生物技术与方法
PCR扩增循环数对细菌群落多样性测序分析的影响
安云鹤1,2,高丽娟1,2,李俊博1,2,田彦捷1,2,王金龙1,2,郑学娟1,2,武会娟1,2
1 北京市理化分析测试中心,北京 100089
2 北京市基因测序与功能分析工程技术研究中心,北京 100094
安云鹤, 高丽娟, 李俊博, 等. PCR 扩增循环数对细菌群落多样性测序分析的影响. 生物工程学报, 2016, 32(8): 1115-1123.
An YH, Gao LJ, Li JB, et al. Influence of PCR cycle number on microbial diversity analysis through next generation sequencing. Chin J Biotech, 2016, 32(8): 1115-1123.
运用高通量测序技术分析复杂样品中微生物群落组成及变化趋势,已经成为目前微生物研究领域的热点之一。本研究以复杂土壤样品和应用范围较广的瘤胃食糜样品为对象,选取20、25和30三个扩增循环数分别对样品的16S rRNA基因的V3区进行扩增,然后进行文库构建和测序。最后通过数据分析比较不同的扩增循环数对细菌多样性测定结果的影响。结果表明,扩增循环数越多,捕获到的细菌数量和种类越多;但并非循环数越多,群落中的微生物组成比例最优。整体来看,当扩增循环数为25时,样品中物种的数量和组成是最优的。
PCR扩增循环数,高通量测序,微生物多样性分析
目前,能够在实验室条件下培养的微生物种类占自然界中微生物总数的不到1%,绝大多数尚未被人类所认识[1]。随着分子生物学技术的发展,PCR指纹技术[2-3]、DNA 文库[4-5]、RAPD[6-7]、ITS[8-9]、DGGE[10-11]等技术相继被广泛应用于微生物群落结构及基因资源的开发利用,从而摆脱了培养的限制。尤其近几年,新一代测序技术以高通量、速度快、低成本等特点迅速发展,在微生物多样性研究领域,正在逐步取代传统的分子生物学方法[12-16],从而进一步丰富和拓展了人们对于未培养微生物的认识。
利用高通量测序技术研究微生物的多样性,对于样品的要求较为严格。其中,目的片段的扩增是进行微生物多样性测序的关键步骤之一。不同的扩增循环数会直接影响最终数据分析中微生物种群的组成和丰度。由此可见,在微生物多样性测序中,环境样品的PCR扩增是非常关键和重要的环节。
尽管之前已有研究对土壤和羊瘤胃食糜样品扩增条件[17-19]的报道,但大多是基于 DGGE方法的,并且很少涉及扩增循环数的优化。目前对高通量测序方法过程中,不同类型样品的PCR扩增条件等方面尚未有系统的研究。本研究以土壤和羊瘤胃食糜样品为对象,对最适PCR扩增循环数进行了比较研究。结果发现,在相同DNA模板量情况下,扩增循环数越多捕获到的微生物种类越多,但会造成优势菌群的过量扩增,从而使其比例变化显著。
1 材料与方法
1.1DNA提取
土壤样品直接称取250 mg进行后续操作;食糜样品离心沉淀并用PBS洗涤后用于与处理土壤样品同样的后续操作。样品中加入 800 L CTAB缓冲液振荡2 m in,加0.3 g 0.1 mm研磨珠和0.1 g 0.5 mm研磨珠,每隔2 m in振荡1次,持续30 m in;70 ℃水浴20 m in后离心收集上清,加入RNAase 37 ℃水浴30 m in以去除样品中的RNA 污染;加入等体积苯酚/氯仿/异戊醇(25:24:1)振荡 30 s至呈白色乳液,离心收集上清;重复萃取步骤1次后,加入0.8倍体积异丙醇,充分混匀,室温放置5-10 m in,放入-20 ℃过夜沉淀DNA;高速离心20 m in,去除上清,用70%乙醇洗涤沉淀,最后风干沉淀并用50-100 L去离子水溶解。最后用BioSpec-nano核酸蛋白测定仪测定DNA的浓度和质量,同时取500 ng DNA进行琼脂糖凝胶电泳。
1.2PCR扩增
选取土壤和食糜样品各 25 ng,分别利用Ex Taq酶对 16S rRNA基因的 V3可变区进行PCR扩增,扩增循环数分别选取 20、25和 30个循环。扩增引物为:Forward:5′-CCTACGGGA GGCAGCAG-3′,Reverse:5′-ATTACCGCGGCT GCTGG-3′。每一条前向引物的5′末端均加入10 bp的一段barcode序列,用于区分不同条件的扩增产物。扩增产物分别命名为S-20c、S-25c、S-30c、C-20c、C-25c和C-30c。
1.3文库构建和测序
将6个扩增产物按照每个100 ng的质量进行混合后,对混合产物进行末端补平加 A、连接接头和少量扩增进行富集。对构建好的文库进行Agilent 2 100和QPCR质检,合格后用于M iseq 2×150 bp的测序。
1.4分析软件
对测序得到的原始数据进行过滤,去掉接头和低质量的测序序列,得到可用于后续分析的高质量数据,并用fastqc软件对数据进行质量评估,使用FLASH软件对高质量数据进行组装,获得更多含barcode的测序序列。再使用QIIME软件根据barcode序列将该组装结果回归样品,并对每个样品的序列数进行统计。然后使用uclust软件根据序列相似性 (阈值为97%) 进行聚类,得到 OTU (操作分类单元) 序列,并进行优化和分类。本研究采用菌群丰富度指数 chao1、ACE,菌群多样性指数Simpson、Shannon和菌群覆盖度指数goods coverage、observed species对菌群进行多样性分析。最后使用 greengenes数据库 (201305版本) 并根据分类学分析结果,分析样品在各分类水平上的物种组成比例情况。
1.5主要试剂和仪器
土壤样品 (简称 S),为山东冬季未经围填的距离围填海区域1.5 km、距表层15 cm的植物根系土壤。样品采集后,冰块运送并于 24 h内处理;羊瘤胃食糜样品 (简称 C),取自立即处死的羊瘤胃胃液,经纱布过滤处理,并立即进行核酸提取;RNAase购自美国NEB公司;0.1 mm和0.5 mm研磨珠购自美国BioSpec公司;CTAB (十六烷基三甲基溴化铵)、苯酚、氯仿、异戊醇、异丙醇、无水乙醇等均购自北京化学试剂公司;Ex Taq DNA聚合酶购自大连宝生物工程有限公司;Truseq DNA library prep kit、Illum ina 2×150 bp测序试剂均购自美国Illum ina公司。高速离心机购自德国 Eppendorf公司;BioSpec-nano核酸蛋白测定仪购自日本岛津公司;PCR仪购自美国Bio-Rad公司;M iseq测序仪购自美国Illum ina公司。
2 结果与分析
2.1土壤和羊瘤胃食糜样品DNA提取
按照材料与方法中DNA提取步骤提取土壤和羊瘤胃食糜样品DNA,然后用BioSpec-nano测定DNA的浓度和质量 (表1)。结果表明,土壤样品的DNA浓度为12.25 ng/L,食糜的提取浓度为11.85 ng/L,A260/280的值均在1.7-2.0之间。琼脂糖凝胶电泳的结果如图 1所示,表明提取的微生物基因组完整,没有RNA等污染。
2.2不同的扩增循环数对测序数据OTU和稀释曲线的影响

表1 土壤和羊瘤胃食糜样品DNA提取浓度Table 1 DNA concentration extracted from soil and chyme sam p les
分别对土壤和食糜样品的 16S V 3区进行20、25和30三个不同扩增循环数的扩增,最终通过数据分析观察不同扩增循环数对最终数据的OTU数目和稀释曲线的影响。实验结果表明,食糜样品的OTU数目随着扩增循环数的增加而显著增加,而土壤样品的OTU数目在扩增循环数为 25时最高;当扩增循环数增加至 30,其OTU数目反而有所下降,但仍较扩增循环数为20时的OTU数目多 (图2A)。S-20c、S-25c、S-30c的OTU数目分别为5 524、11 852和8 172;C-20c、C-25c、C-30c的OTU数目分别为1 737、2 663和7 233。每个样品的稀释曲线基本都呈逐渐平缓的趋势 (图2B),说明测序数据基本能够覆盖目前状态下的微生物种类。
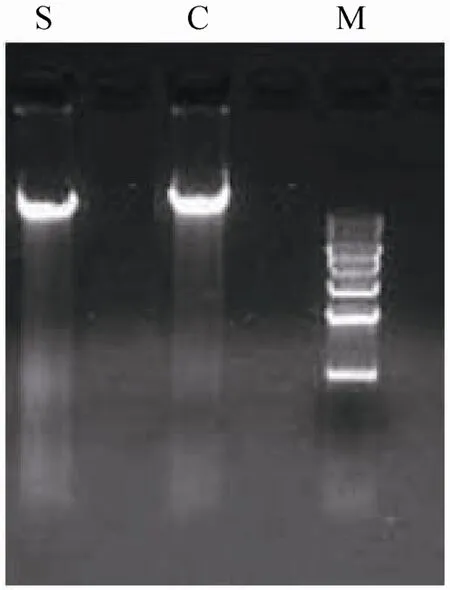
图1 土壤和食糜样品DNA胶图Fig. 1 Electrophoretogram of DNA extracted from soil and chyme samples. S: soil; C: chyme; M: 1 kb DNA marker.
2.3不同扩增循环数对微生物数量和种类的影响
实验结果表明,随着扩增循环数的增加,微生物数量都呈上升趋势。但土壤样品和食糜样品的情况略有不同,土壤样品在扩增至25个循环时,微生物数量最多,当循环数增加至 30时,微生物的数量改变不大。如图3所示,S-20c、S-25c和S-30c获取的微生物数目分别是:49 475、169 257和147 895。而对于食糜样品,微生物的数量随着扩增循环数的增加而呈逐渐上升的趋势,但扩增循环数为30时,较扩增循环数为25时的微生物数量上升缓慢。如图 3所示,
C-20c、C-25c和C-30c获取的微生物数目分别是:17 631、44 788和56 492。
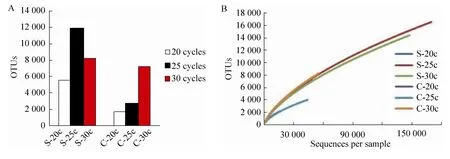
图2 不同扩增循环数对测序数据OTU和稀释曲线的影响Fig. 2 Effect of different amplification cycles on OTU and Rarefaction curves. (A) Effect of different amplification cycles on OTU in soil and chyme samples. (B) Effect of different amplification cycles on Rarefaction curves in soil and chyme samples.
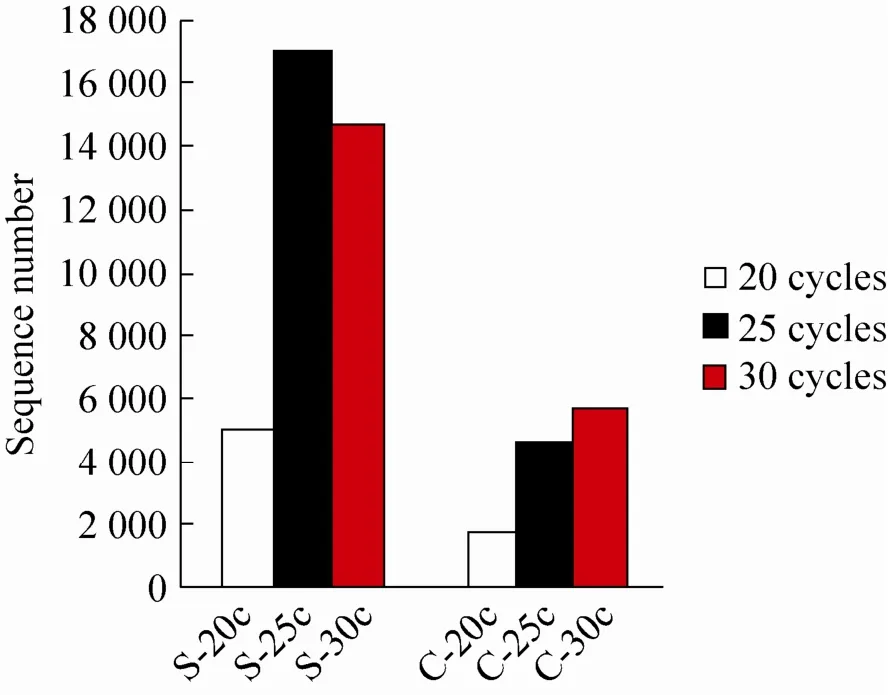
图3 不同扩增循环数对微生物数量的影响Fig. 3 Effect of different amplification cycles on m icrobial population.
从微生物多样性分析的结果来看,物种较为丰富的土壤样品,当扩增循环数为25时,其微生物种类为16 263,基本达到饱和程度;当扩增循环数为30时,样品中的微生物种类为14 188,微生物的种类没有增加,反而有一定程度的降低 (表 2)。而对于食糜样品,随着扩增循环数的增加,捕获到的微生物种类呈递增趋势。如表2所示,C-20c/25c/30c捕获的微生物种类分别是2 202、3 908和8 030。
在门水平上对样品中微生物的群落组成进行分析的结果表明,并非扩增循环数越多,样品中微生物的群落组成越丰富。对于物种含量较丰富的土壤样品来说,3个不同的扩增循环数对于丰度较高菌门的微生物捕捉是一致的,但对丰度较低菌门的微生物捕捉差别较大。当扩增循环数为25时,微生物的种类最多;而当扩增循环数为30时,微生物的种类反而会减少,尤其是丰度较低的菌门微生物,很多在扩增循环数为 25时捕捉到的微生物种类缺失,如BHI80-139、H-178、LD1、AncK 6等,组成比例也相应地变化较大 (图4A)。而对于物种含量较少的食糜样品,总体来看,随着扩增循环数的增加,物种含量也相应增多。但当扩增循环数为30时,其优势菌门的微生物含量比例变化较大 (图4B)。因此,从2种样品的分析结果来看,当扩增循环数为25时,物种的丰富度和群落组成比例更为合理。
3 讨论
高通量测序技术通过对高质量样品的快速测序,以一种更为便捷经济的方式,可以获得样品中全面、准确、无偏的数据信息。也正因如此,利用高通量测序技术的微生物多样性测序[12-14,16]和宏基因组测序[15,20-21]越来越得到研究者的重视和青睐,其应用范围也越来越广。
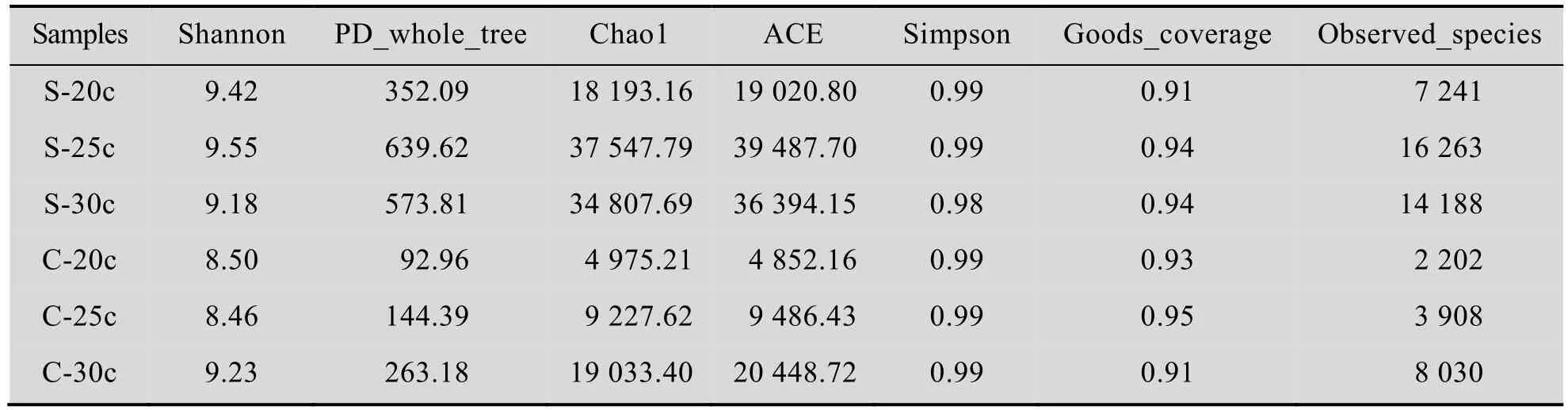
表2 不同扩增循环数对微生物种类的影响Tab le 2 Effect of different am p lification cycles on m icrobial species
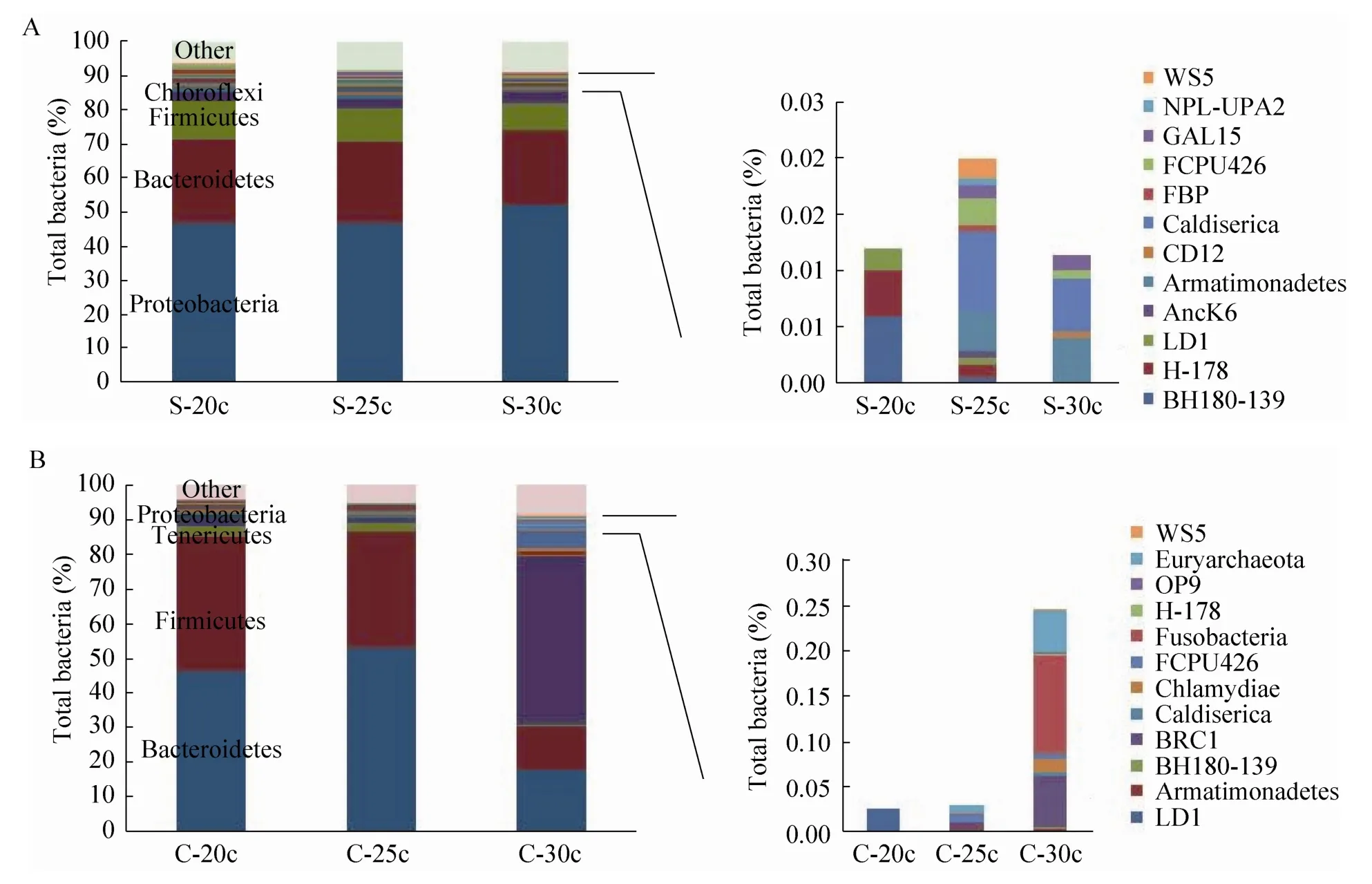
图4 不同扩增循环数对微生物门水平群落组成的影响Fig. 4 Effect of different amplification cycle number on m icrobial community composition. (A) Effect of different amplification cycle number on m icrobial community composition in soil samples. (B) Effect of different amplification chloroflexi cycle number on m icrobial community composition in chyme samples.
高通量测序技术对样品的要求较高,不合格的样品质量将造成后续数据分析的巨大偏差。土壤中富含腐殖酸等化合物[22],因此提取的DNA纯度往往不能令人满意,影响后续PCR反应和DNA测序[23]。存在于动物和人体中的食糜样品,无论是取自胃部还是肠道,其中所含微生物种类对于健康[24]、养殖[13]、发酵[25]等领域均具有重要的理论意义。在此研究中,我们选取环境样品中较难处理的土壤样品和应用范围较广的食糜样品,旨在得到一套可用于指导高通量测序前期样品DNA提取及扩增条件的技术参数。
从DNA的提取结果来看,不论是土壤样品,还是食糜样品,DNA的质量均符合要求。我们选取25 ng DNA作为模板成功地进行后续PCR扩增,然后将扩增产物用于建库、测序及数据分析。从数据产生的OTU来看 (图2A),土壤样品获得的OTU数目远远多于食糜样品,这暗示土壤中所含的细菌种类较多,而食糜样品中种类较少。此外,土壤中所含的细菌数量也远远高于食糜样品 (图 3)。这些数据均表明,土壤样品中微生物的复杂程度远远高于食糜样品。因此在扩增循环数的选择上也有所不同。
就土壤样品而言,当扩增循环数达到25时,其细菌种类和数量均达到最高值。再增加循环数,二者均会有所降低 (图 3和表 2)。同时,从群落组成的结果来看,扩增循环数为25时,土壤中微生物的组成比例最优 (图4A)。不但保持扩增循环数为20时的优势菌门种类和比例,同时最大程度增加了劣势菌门的种类。而当扩增循环数为30时,不但会丢失低扩增循环数下捕捉到的劣势菌,如BHI80-139,并且增加的劣势菌种类也远远不及扩增循环数为 25时的结果。
在食糜样品中,随着扩增循环数的增加,细菌种类和数量都呈上升趋势。其中,细菌的种类随着扩增循环数的增加,上升趋势显著 (表2);而扩增循环数为30时,细菌的数量只比扩增循环数为25时的有小幅提升 (图3),说明当扩增循环数为25时,细菌的数量已经趋于饱和。这些数据表明,从扩增循环数25增加至30,扩增的产物总量变化不大,而种类发生了变化。从食糜样品的群落组成来看,当扩增循环数为30时,4个优势菌,如拟杆菌门Bacteroidetes、厚壁菌门Firmicutes、软壁菌门 Tenericutes、变形菌门Proteobacteria,其比例组成较扩增循环数为20和25时均有大幅变化。而劣势菌得到了大幅扩增,不但种类更多,同时组成比例也相应增大 (图4B)。
因此,对于食糜这类微生物含量较低的样品而言,如果要关注优势菌的种类和比例,选择扩增循环数为25会更有利于结果分析;如果要关注其中的劣势菌,则选择扩增循环数为 30会更有利。对于土壤这种微生物含量丰富的样品,扩增循环数选择 25是最优的。这与利用DGGE法分析微生物多样性,所选用的扩增循环数有所不同。DGGE技术主要是利用梯度变性胶来分离DNA片段。其原理是不同碱基组成的DNA双螺旋,在线性梯度变性剂中发生解链的位置不同,从而造成在变性胶中涌动的位置不同而达到分离的目的。因此,在进行电泳前,样品的扩增产物要满足两个条件:一个是尽可能保持扩增产物的稳定性以增加电泳时间,达到精确分离的目的;一个是尽量增加扩增产物量以保证每个分离条带的可辨性。因此,DGGE一般会在引物的5′端添加30 bp左右的GC夹子,同时扩增循环数一般不低于 30个循环。DGGE方法中关于PCR条件的优化有很多研究报道,但主要集中在PCR技术的选择上[17-19,26],如普通PCR、降落PCR、巢式PCR或其他技术改进。这一方面与GC夹子的引入有关,一方面与扩增产物的量必须达到相对较高的水平有关。
不论是利用高通量测序技术还是DGGE技术,在分析微生物的多样性组成时,都涉及DNA样品的PCR扩增,即均需要对样品进行富集。一般情况下,扩增循环数越少,越能反应样品的真实情况;但扩增循环数越少,能够捕捉到的样品信息也会越少。为了平衡样品的真实性和信息的全面性,我们比较了20、25、30三个扩增循环数对微生物群落组成的影响。从分析结果来看,无论是土壤样品还是食糜样品,当扩增循环数为25时,细菌的群落组成与扩增循环数为20时的群落组成最接近,即最接近真实状态,同时其在物种多样性上较扩增循环数为20时的物种多样性更高。如果要最大程度地反映样品的真实性,可以选择宏基因组测序技术[15,20-21],即直接对环境样品中的DNA进行打断和测序,不需要进行PCR扩增。这样在进行数据分析时,可以通过拼接 16S rDNA的全长基因实现微生物的种类鉴定。
REFERENCES
[1] Wommack KE, Nasko DJ, Chopyk J, et al. Counts and sequences, observations that continue to change our understanding of viruses in nature. J M icrobiol, 2015, 53(3): 181-192.
[2] Secundo de Souza AI, Freitas Neto OC, Batista DF,et al. ERIC-PCR genotyping of field isolates of Salmonella enterica subsp. enterica serovar Gallinarum biovars Gallinarum and Pullorum. Avian Pathol, 2015, 44(6): 1-20.
[3] M o QH, Wang HB, Tan H, et al. Optim ization and head-to-head comparison of M ISSR-PCR,ERIC-PCR, RAPD and 16S rRNA evolutionary clock for the genotyping of Vibrio cholerae isolated in China. Indian J Med M icrobiol, 2015, 33(4): 516-523.
[4] Dewhirst FE, K lein EA, Bennett M L, et al. The feline oral m icrobiome: a provisional 16S rRNA gene based taxonomy w ith full-length reference sequences. Vet M icrobiol, 2015, 175(2/4): 294-303.
[5] Hotta F, Eguchi H, Naito T, et al. Achromobacter buckle infection diagnosed by a 16S rDNA clone library analysis: a case report. BMC Ophthalmol,2014, 14: 142-148.
[6] Ren W, Zhao H, Shao W, et al. Identification of a novel phenamacril-resistance related gene by cDNA-RAPD method in Fusarium asiaticum. Pest M anag Sci, 2015.
[7] Sadiq FA, Li Y, Liu T, et al. A RAPD based study revealing a previously unreported w ide range of mesophilic and thermophilic spore formers associated w ith m ilk powders in China. Int J Food M icrobiol, 2015, 217: 200-208.
[8] Choi IW, Kim HY, Quan JH, et al. M onitoring of fasciola species contam ination in water dropwort by cox1 m itochondrial and ITS-2 rDNA sequencing analysis. Korean J Parasitol, 2015, 53(5): 641-645.
[9] Marques S, Huss VA, Pfisterer K, et al. Internal transcribed spacer sequence-based rapid molecular identification of Prototheca zopfii and Prototheca blaschkeae directly from milk of infected cows. J Dairy Sci, 2015, 98(5): 3001-3009.
[10] Flórez AB, Mayo B. Diversity and dynam ics of antibiotic-resistant bacteria in cheese as determ ined by PCR denaturing gradient gel electrophoresis. Int J Food M icrobiol, 2015, 214: 63-69.
[11] El-Sayed WS1, Ouf SA2, Mohamed AA3. Deterioration to extinction of wastewater bacteria by non-thermal atmospheric pressure air plasma as assessed by 16S rDNA-DGGE fingerprinting. Front M icrobiol, 2015, 6: 1098-1110.
[12] Xun W, Zhao J, Xue C, et al. Significant alteration of soil bacterial communities and organic carbon decomposition by different long-term fertilization management conditions of extremely low-productivity arable soil in South China. Environ M icrobiol, 2016, 18(6): 1907-1917.
[13] Jami E, Israel A, Kotser A, et al. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J, 2013, 7(6): 1069-1079.
[14] Kautz S, Rubin BE, Russell JA, et al. Surveying the microbiome of ants: comparing 454 pyrosequencing w ith traditional methods to uncover bacterial diversity. Appl Environ M icrobiol, 2013, 79(2): 525-534.
[15] Bulgarelli D, Garrido-Oter R, Münch PC, et al. Structure and function of the bacterial root microbiota in w ild and domesticated barley. Cell Host M icrobe, 2015, 17(3): 392-403.
[16] Dassi E, Ballarini A, Covello G, et al. Enhanced m icrobial diversity in the saliva microbiome induced by short-term probiotic intake revealed by 16S rRNA sequencing on the IonTorrent PGM platform. J Biotechnol, 2014, 190: 30-39.
[17] Yin QY, Guo XL, Zhao MQ, et al. Optim ization of V3 region PCR reaction system w ith soil m icrobial total DNA as template. J Henan Agricul Sci, 2012,41(1): 65-68.
殷全玉, 郭夏丽, 赵铭钦, 等. 土壤微生物总DNA的V3可变区PCR反应体系优化. 河南农业大学, 2012, 41(1): 65-68.
[18] Wang M J, Cao YS, Hu YX, et al. Investigating the bacterial diversity of chicken respiratory tract byPCR-DGGE. China Feed, 2013, 4(9): 26-28.
汪梦娟, 曹银生, 胡艳霞, 等. 应用 PCR-DGGE分析肉鸡呼吸道中细菌多样性. 中国饲料, 2013,4(9): 26-28.
[19] Jiang HY, Chen ZL, Zhao GH, et al. Investigating the diversity of lactic acid bacteria in Tibetan traditional fermented dairy products by PCR-DGGE. Food Science, 2014, 35(1): 167-173.
蒋厚阳, 陈芝兰, 赵国华, 等. PCR-DGGE法分析西藏传统发酵乳制品中乳酸菌的多样性. 食品科学, 2014, 35(1): 167-173.
[20] Bhatia S, Batra N, Pathak A, et al. Metagenom ic analysis of bacterial and archaeal assemblages in the soil-mousse surrounding a geothermal spring. Genom Data, 2015, 5: 195-200.
[21] Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women w ith normal,impaired and diabetic glucose control. Nature,2013, 498(7452): 99-103.
[22] Courtois S, Frostegård A, Göransson P, et al. Quantification of bacterial subgroups in soil: comparison of DNA extracted directly from soil or from cells previously released by density gradient centrifugation. Environ M icrobiol, 2001, 3(7): 431-439.
[23] Hurt RA, Qiu X, Wu L, et al. Simultaneous recovery of RNA and DNA from soils and sediments. Appl Environ M icrobiol, 2001, 67(10): 4495-4503.
[24] Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germ free m ice. ISME J, 2013, 7(4): 880-884.
[25] Yin YY, Liu YJ, Zhu WY, et al. Effects of acarbose addition on ruminal bacterial m icrobiota,lipopolysaccharide levels and fermentation characteristics in vitro. Asian-Australas J Anim Sci,2014, 27(12): 1726-1735.
[26] Wang YF, Zhang FQ, Gu JD. Improvement of DGGE analysis by modifications of PCR protocols for analysis of m icrobial community members w ith low abundance. Appl M icrobiol Biotechnol, 2014,98(12): 5655-5663.
(本文责编 陈宏宇)
Influence of PCR cycle number on microbial diversity analysis through next generation sequencing
Yunhe An1,2, Lijuan Gao1,2, Junbo Li1,2, Yanjie Tian1,2, Jinlong Wang1,2, Xuejuan Zheng1,2, and Huijuan Wu1,2
1 Beijing Center For Physical and Chemical Analysis, Beijing 100089, China
2 Beijing Engineering Technique Research Center for Gene Sequencing & Function Analysis, Beijing 100094, China
Using of high throughput sequencing technology to study the microbial diversity in complex samples has become one of the hottest issues in the field of microbial diversity research. In this study, the soil and sheep rumen chymesamples were used to extract DNA, respectively. Then the 25 ng total DNA was used to amplify the 16S rRNA V3 region w ith 20, 25, 30 PCR cycles, and the final sequencing library was constructed by m ixing equal amounts of purified PCR products. Finally, the operational taxonom ic unit (OUT) amount, rarefaction curve, m icrobial number and species were compared through data analysis. It was found that at the same amount of DNA template, the proportion of the community composition was not the best w ith more numbers of PCR cycle, although the species number was much more. In all, when the PCR cycle number is 25, the number of species and proportion of the community composition were the most optimal both in soil or chyme samples.
October 23, 2015; Accepted: January 6, 2016
Huijuan Wu. Tel: +86-10-58711817; Fax: +86-10-58717638; E-mail: sunnywhj@126.com
PCR cycle number, high throughput sequencing, m icrobial diversity analysis
Supported by: Beijing Academy of Science and Technology 2015 Youth Backbone Project (No. 201522).
北京市科学技术研究院2015年青年骨干项目 (No. 201522) 资助。
网络出版时间:2016-03-17 网络出版地址:http://www.cnki.net/kcms/detail/11.1998.Q.20160317.1018.001.html
猜你喜欢
动物营养学报(2022年7期)2022-08-09
中国医学物理学杂志(2022年6期)2022-07-01
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
收藏界(2018年1期)2018-10-10
创新作文(小学版)(2018年31期)2018-05-16
摄影之友(影像视觉)(2017年1期)2017-07-18
中国发展观察(2017年8期)2017-04-26
小学阅读指南·低年级版(2016年6期)2016-05-14
中国当代医药(2015年33期)2015-03-01
