
黑曲霉组学研究进展
2016-09-13 08:36隋雨菲欧阳立明鲁洪中庄英萍张嗣良
生物工程学报 2016年8期
隋雨菲,欧阳立明,鲁洪中,庄英萍,张嗣良
华东理工大学 生物反应器工程国家重点实验室,上海 200237
综 述
黑曲霉组学研究进展
隋雨菲*,欧阳立明*,鲁洪中,庄英萍,张嗣良
华东理工大学 生物反应器工程国家重点实验室,上海 200237
隋雨菲, 欧阳立明, 鲁洪中, 等. 黑曲霉组学研究进展. 生物工程学报, 2016, 32(8): 1010-1025.
Sui YF, Ouyang LM, Lu HZ, et al. Progress in omics research of Aspergillus niger. Chin J Biotech, 2016, 32(8): 1010-1025.
黑曲霉作为重要的工业发酵菌株,被广泛用于多种有机酸和工业用酶的生产。随着组学技术的日益发展和成熟,黑曲霉的基因组、转录组、蛋白质组、代谢组等组学数据不断增长,宣告着黑曲霉生物过程研究大数据时代的到来。从单一组学的数据分析、多组学的比较到以基因组代谢网络模型为中心的多组学整合研究,人们对黑曲霉高效生产机制的理解不断深入和系统,这为通过遗传改造和过程调控对菌株的生产性能进行理性的全局优化提供了可能。本文回顾和总结了近年来黑曲霉的组学研究进展,并提出黑曲霉组学研究未来的发展方向。
黑曲霉,基因组,转录组,蛋白质组,代谢组,组学整合,基因组规模代谢网络模型
黑曲霉 Aspergillus niger是一种丝状子囊真菌,在环境中十分常见[1]。在自然生长状态下,黑曲霉分泌大量多种用途的酶降解环境中的生物聚合物从而获得营养。黑曲霉生产的工业酶在淀粉加工、发酵、酿造、饮料生产动物饲料和造纸行业等领域发挥了巨大的作用。此外,黑曲霉还可以作为异源蛋白的生产宿主及生产柠檬酸和葡萄糖酸的细胞工厂,因其具有高产、高分泌、高安全性等优点而被广泛地应用于发酵工业中[2]。黑曲霉基因组序列和注释信息的公布[1-4],大大推进了后续的各种组学研究,使人们能够更加深入分析这一重要黑色真菌的代谢潜力,以及应用现代生物技术进一步提高它的生产能力[5]。
1 黑曲霉基因组学进展
黑曲霉基因组序列的发表为其他组学的研究和生物技术的应用提供了新的平台[5]。新一代测序技术 (Next Generation Sequencing,NGS)为快速获得多种基因组序列提供了高效的工具[3],为基因组和转录组测序和分析带来了革命性的突破。目前,已经有 4株黑曲霉完成了全基因组测序 (表 1),分别为野生型菌株 ATCC 9029 和高产柠檬酸菌株ATCC 1015,经过传统诱变方法得到的高产酶菌株CBS 513.88,以及无孢子型糖化酶工业菌株SH2。
2000年,荷兰帝斯曼公司率先开启了黑曲霉的全基因组测序工作,对产酶菌株 CBS 513.88进行基因组测序。结果于2007年发表。并对基因组序列进行了较完善的功能注释,从14 165个预测的结构基因中识别了6 506个与代谢调控、细胞转运和蛋白质降解有关的开放阅读框。在基因组序列和注释基础上,他们重新构建了包含 1 069个特异性反应的黑曲霉代谢网络模型,很好地解释了黑曲霉高效生产葡萄糖淀粉酶的机制,充分体现了黑曲霉代谢网络的多样性[2]。
第二个进行全基因组测序的黑曲霉菌株是实验室常用的ATCC 9029。由Integrated Genom ics完成测序并于2005年发表,但是仅构建了高度碎片化的序列草图 (9 510个重叠群(Contigs)),没有公开发布基因注释或者基因预测结果[6]。
The Joint Genome Institute (JGI) 在美国能源部资助下对柠檬酸高产菌株ATCC 1015进行测序[5],通过比对发现其97%的基因序列与CBS 513.88一致,有0.8 Mb新序列,并存在较多的基因组重排、缺失、菌株特异性水平基因转移现象。而且两种菌株之间单核苷酸多态性(SNP)水平很高 (平均:7.8 SNPs/kb;最大:160 SNPs/kb)。相比于其他曲霉,黑曲霉具有大量特异性蛋白参与蛋白质、碳水化合物、脂肪酸以及类异戊二烯等次级代谢途径的代谢,反映了黑曲霉作为细胞工厂的多功能性。不过,不同曲霉属丝状真菌间与细胞运输、蛋白分泌和蛋白寿命有关的特异性蛋白数量基本不变[2]。
2014年华南理工大学对无孢子糖化酶高产菌株SH2进行基因组测序,识别了约11 517个开放阅读框。通过比较基因组学分析了黑曲霉SH2和CBS 513.88菌株的不同,发现SH2菌株缺失与无性孢子形成有关的基因 (PrpA)。另外细胞壁多糖合成和细胞壁完整性信号通路相关基因出现了序列变异。比较代谢基因组学表明黑曲霉具有较高的代谢特异性,含有超过1 100个编码酶的特异性基因[3]。
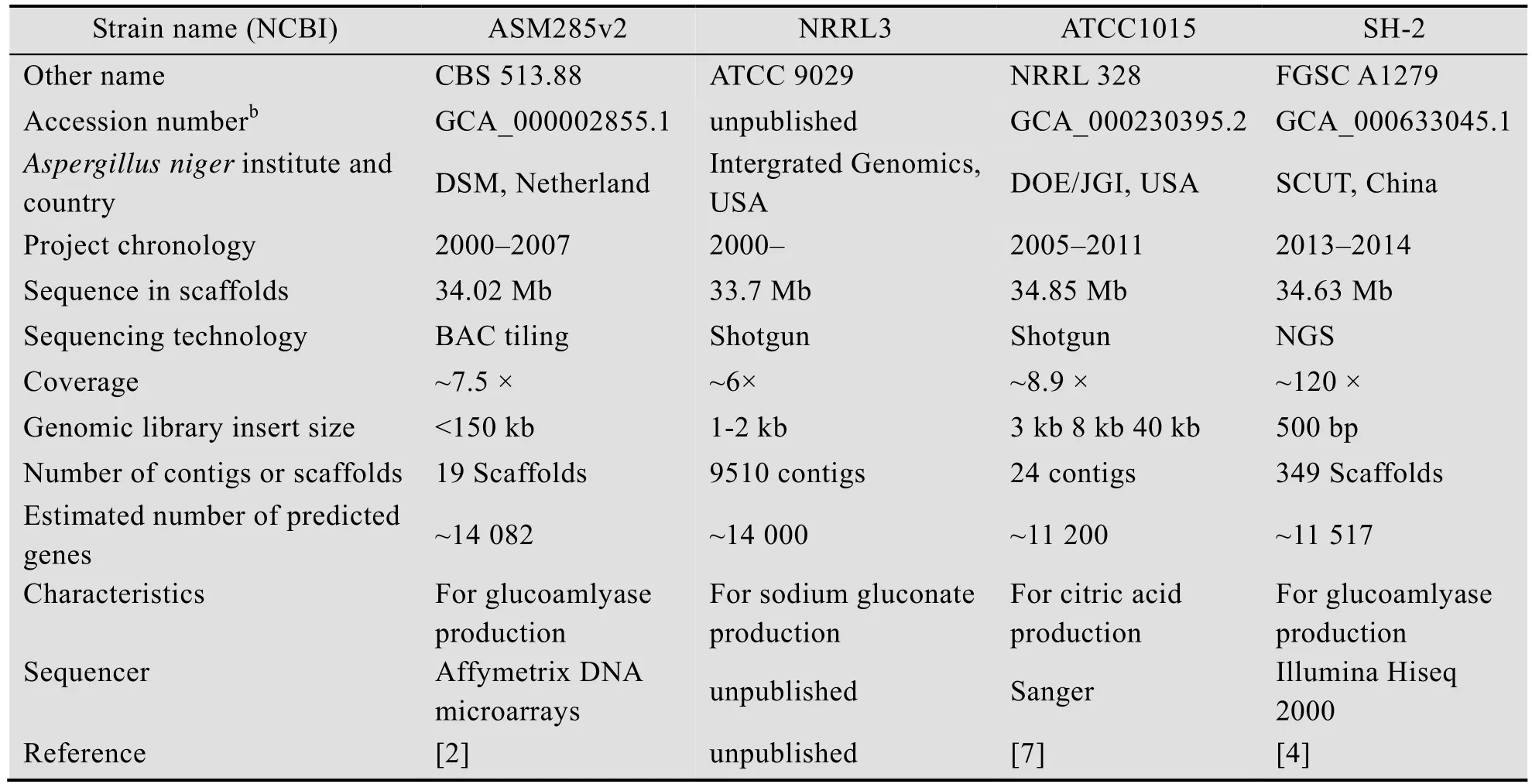
表1 已发表的黑曲霉基因组aTab le 1 The pub lished genom es of Aspergillus niger strains
越来越多的曲霉属基因组测序是开展比较基因组学研究的坚实基础。基因组序列及注释信息和基因组规模代谢网络模型的构建使得其他单一组学的数据分析以及多组学整合成为可能。
2 黑曲霉转录组学进展
全转录组层次的数据分析可以使研究者更加精确地评估细胞表型与基因表达的关系,加深对细胞代谢的理解[7]。随着测序技术的发展,近年来越来越多转录组研究采用新一代的高通量转录组测序技术 (RNA sequencing,RNA-seq)。相比于基因芯片,RNA-Seq技术能够获得新转录本,可变剪切等更多的基因结构和功能信息,提高测序结果的准确性。测序技术的日益进步推动了转录组学的发展及其在代谢工程领域的应用。
2.1用转录组学方法研究黑曲霉高产酶机制
黑曲霉作为生产工业用酶的重要生产菌株,深入研究其高效产酶机制和分泌机制具有重要的现实意义。目前,转录组学分析广泛地应用于分析不同环境变量,不同产酶水平菌株之间产酶量差异的关键因素,寻找起到关键调控作用的基因或基因簇,为后续的菌种改造提高黑曲霉产酶性能提供潜在的靶点。
2011年Andersen等[8]对产酸株ATCC 1015和产酶株CBS 513.88进行相同培养条件下的转录组学比较分析,结果表明产酶菌株 CBS 513.88的参与糖酵解途径,TCA循环和氨基酸代谢有关的基因表达上调,尤其与苏氨酸、丝氨酸和色氨酸这 3个氨基酸合成途径相关的基因上调显著,而这些氨基酸在葡萄糖淀粉酶 (又名糖化酶,Glucoamylase,GlaA),tRNA合成酶,蛋白质转运子中含量较高;ATCC 1015中参与生物氧化和电子传递链、碳水化合物和有机酸转运的基因显著上调。代谢流分析结果表明CBS 513.88中3种氨基酸生物合成途径的通量至少是ATCC 1 015的2倍,与转录组的结果吻合。
为了从全基因组角度分析黑曲霉蛋白分泌途径的转录调控网络,Kwon等[4]发现相比于野生型,glaA过表达菌株中GlaA的mRNA和胞外GlaA水平显著提高,同时还伴随着772个基因转录水平提高,815个基因转录水平下调。GO (Gene ontology) 富集分析发现,上调基因主要涉及 4类功能:内质网 (ER) 跨膜运输、蛋白质糖基化、囊泡运输和离子平衡。为通过遗传改造手段增强目的蛋白的生产能力提供了有力的参考信息。
Jørgensen等[9]设计了一个新的细胞截留装置,可以在高流速下实现几乎全部细胞的截留。转录组分析用于探索低比生长速率下产物合成的潜力。比生长速率接近0时 (<0.005 h-1),比生长得率稳定在最小值 (0.20 g干重/g麦芽糖)。选择保留培养3个生理学特征明显的阶段 (0、2、8 d) 进行转录组学分析。结果表明,比生长速率接近于零时,细胞表现出从生长阶段到繁殖阶段的过渡,编码分泌型水解酶和细胞周期蛋白的基因下调表达,许多编码分泌型富含半胱氨酸的小蛋白和次级代谢产物的基因上调表达。通过以上结果,预测比生长速率接近 0条件下培养黑曲霉更适于生产次级代谢物和富含半胱氨酸的蛋白。为研究优化黑曲霉中酶蛋白的生产条件提供新的方向。
2.2转录组学揭示黑曲霉在不同碳源条件下的转录响应
转化复杂的纤维素和半纤维素成为菌体可利用的简单碳源是生产二代生物燃料的主要挑战[9]。真菌是生产植物多糖降解酶类,如纤维素酶和半纤维素酶的重要来源。这些酶在生物燃料的制造过程中有重要应用,也是生物燃料制造成本中的重要部分。目前的转录组研究表明,碳源的种类和水平可以极大地影响黑曲霉中多糖降解酶类的生产。
JØrgensen等[10]在恒化培养且比生长速率相同的条件下,比较木糖和麦芽糖分别作为碳源时黑曲霉的转录组情况,发现以麦芽糖作为底物时,胞外酶的生产速率是木糖作为底物时的 3倍,且蛋白分泌增强。转录组中显著上调的基因中有90个以上与蛋白分泌途径相关。这些基因的功能涉及蛋白质从核糖体运输到内质网、折叠、N端糖基化、质量控制、囊泡包装以及从内质网到高尔基体之间的运输等过程。另外还有大约890个基因的表达差异倍数小于2倍,包括分泌途径和中心代谢途径的基因。在不同条件下表达差异较小是细胞基本功能基因的特点。
Yuan等[11]研究表明AmyR是黑曲霉中重要的麦芽糖依赖型转录因子,它的减少使得胞外水解酶 (α-淀粉酶 (AamA),α-葡萄糖苷酶(AgdA和 AgdB),以及葡萄糖淀粉酶 (GlaA))酶量降低,这些酶能够将麦芽糖转化为葡萄糖。胞外水解酶的减少导致可利用的葡萄糖减少从而产生一种信号使得葡萄糖转运子也下调表达。Vongsangnak等[12]的转录组学和生理学数据证实了Yuan的结论,表明黑曲霉利用AmyR转录因子调控系统而非 MAL调节系统激活胞外水解酶类,如葡萄糖淀粉酶 (GlaA),从而提高菌体对麦芽糖的利用。MAL调节系统在米曲霉中存在,但在黑曲霉中不存在[12]。
降解植物细胞壁多糖的酶主要分为 3类:葡萄糖苷水解酶 (GH)、糖酯酶 (CE) 和多糖裂解酶 (PL)[13]。Souza等[14]考察了以甘蔗渣作为碳源时的转录组,与以葡萄糖为碳源时的转录组相比,黑曲霉中18个纤维素酶基因和21个半纤维素酶基因 (各占黑曲霉基因组中预测为纤维素酶和半纤维素酶基因数量的 58%) 表达发生显著上调。Pullan等[15]研究黑曲霉在速生柳 (一种能源植物) 诱导下的基因表达谱,并且与之前研究过的小麦秸秆诱导的基因表达谱进行比较,发现这两种情况下编码糖活性酶(Carbohydrate active enzyme, CAZyme) 的基因表达都大幅上调。黑曲霉受小麦秸秆诱导时编码GH62 (阿拉伯呋喃糖酶) 和CE1 (阿魏酸酯酶) CAZyme家族酶的基因表达水平更高;而受柳树诱导时,编码属于GH5家族的纤维素内切酶的两个基因表达水平更高。
碳饥饿条件能够诱导黑曲霉中部分多糖降解酶类的分泌,使其在严峻的条件下通过降解自身细胞壁或较难利用的碳源来维持细胞生长。Delmas等[13]发现暴露于小麦秸秆24 h后,编码植物细胞壁降解酶的基因表达量很高 (约占全部mRNA的20%),然而葡萄糖对降解酶类的基因表达有抑制作用。研究发现新的糖酯酶和表面相互作用蛋白,可能具有降解植物细胞质酶的功能。为研究降解酶基因的诱导顺序,作者构建了转录因子X lnR (木聚糖降解激活子,调节木糖诱导的糖苷水解酶和糖酯酶),CreA(碳代谢物阻遏因子) 基因分别缺失的两个突变株。发现碳源饥饿条件可以诱导黑曲霉释放一小部分碳源降解酶类。这些酶起到侦查复杂的植物多糖的作用,降解细胞壁释放少量结构简单的糖和低聚糖,从而进一步诱导黑曲霉表达完整的降解酶系。碳源饥饿培养条件诱导黑曲霉分泌对植物多糖具有活性的 CAZyme,这一现象和诱导机制在 Munster等[16]的工作中也得到了证实。碳源饥饿不仅诱导黑曲霉分泌多糖降解酶类,也对细胞形态的发育分化产生极大影响。Nitsche等[17]首次从系统水平分析黑曲霉液体发酵时碳源饥饿引起的生理学、形态学和转录水平改变。饥饿条件引起了复杂的形态改变和细胞差异,形成空菌丝和分生孢子,而未分支的细小菌丝获得二次生长取代老菌丝。与对数生长期的转录组相比,碳源饥饿条件下 3个取样点 (16、60和140 h) 中至少有1个取样点中52%的基因 (7 292个) 发生了差异表达。其中,自噬和分生孢子形成途径的基因在转录水平上受到明显的诱导。转录组和分泌蛋白质组的数据表明,饥饿条件高度诱导编码水解酶的基因转录使得水解酶 (包括几丁质酶、葡聚糖酶、蛋白酶、磷脂酶等) 分泌水平提高,从而有助于降解细胞壁为菌体的生长提供碳源。
黑曲霉中转录因子A raR调控降解植物多糖主要的纤维素/半纤维素酶基因系统。Souzaa等[18]研究了黑曲霉中受到转录激活因子(X lnR,A raR) 调控的基因群。应用野生株、黑曲霉转录因子 AraR (L-阿拉伯糖合成途径特异性调控子) 和X lnR (木聚糖/纤维素降解系统特异性调控子) 基因缺失突变株,比较它们分别以D-木糖/L-阿拉伯糖单糖混合物和蒸汽爆破的甘蔗渣作为碳源时的转录组差异。确认了受到XlnR和AraR调控的基因群,它们的功能主要涉及纤维素和半纤维素的降解和酶蛋白的转运。
不同的碳源条件对黑曲霉表达多糖降解酶类具有不同的激活和抑制作用,因此寻找适合工业生产的碳源不仅可以有效地提高酶的产量还能够降低生产成本。
2.3转录组学分析黑曲霉孢子萌发时的转录响应
黑曲霉在不同发育状态下转录组差异很大,转录组学分析能够揭示孢子从休眠到萌发以及萌发过程中胞内基因表达谱的变化。
Novodvorska等[19]首次采用RNA-Seq技术研究了黑曲霉不同发育状态下的转录组变化,研究结果揭示出休眠孢子与萌发孢子的转录组差异很大,而主要的改变发生在孢子萌发的第一个小时内。休眠的打破与参与蛋白质合成,RNA转录和呼吸代谢等基因转录水平的提高有关。休眠孢子的转录组含有参与糖异生作用和乙醛酸循环的基因,有利于黑曲霉在饥饿条件诱导的分生孢子形成过程中利用非糖类碳源获得能量,或用于休眠过程中维持菌体的生命活动。此外,休眠孢子的转录组含有重要的反义转录产物,推测这些反义转录产物的表达调控功能与孢子从休眠过渡到完全萌发状态密切相关。
Leeuwen等[20]研究黑曲霉孢子萌发的前 8小时内抗真菌物质纳他霉素对孢子转录组的影响。当分别用3和10 µmol/L纳他霉素处理黑曲霉孢子后,菌丝的两极分化、芽管形成和有丝分裂受到抑制,10 µmol/L时菌体的同向生长也受到影响。纳他霉素阻碍了细胞内甘油和葡萄糖浓度的提高,显著影响了萌发前 2小时转录谱的改变,在这一阶段,与转录、蛋白合成、能量和细胞循环、DNA修饰相关的基因表达明显上调。在3和10 µmol/L纳他霉素条件下分别发现280和2 586个差异表达基因。参与麦角固醇合成的基因下调表达,但是参与内吞作用,相容性溶质代谢、编码保护蛋白的基因上调表达,是黑曲霉在内吞作用被纳他霉素抑制时的应激反应。
3 黑曲霉蛋白质组学研究
黑曲霉中蛋白的生产和分泌是一个十分复杂的过程,且同源和异源蛋白的产量差异很大,同源蛋白的产量是异源蛋白产量的 10-100倍[21]。说明真菌在表达和分泌异源蛋白过程中遇到瓶颈。为从全局水平研究外源蛋白在黑曲霉中表达和分泌受限的机制,需要借助现代组学技术研究环境、基因表达调控与蛋白合成及转运之间的复杂关系。真菌蛋白质组学技术的发展,为工业菌种改造和改进工业发酵过程提供新的途径。
3.1培养基对黑曲霉胞内蛋白质组的影响
前文已提到,碳源的种类和水平极大地影响了黑曲霉的产酶能力。除了碳源,培养基中的其他成分也可能显著影响黑曲霉的蛋白质组。SØrensen等[22]采用蛋白质组学分析表明在含有硝酸盐和淀粉的培养基中添加乳酸盐能够显著提高黑曲霉产伏马菌素B2 (Fumonisin B2,FB2) 的产量。在分别含有3%淀粉,3%淀粉和3%乳酸盐,3%乳酸盐这3种培养条件下黑曲霉的蛋白质组差异很大。共识别到56个与中心代谢途径相关的差异表达蛋白质,其中大部分涉及磷酸戊糖途径、丙酮酸代谢、三羧酸循环、铵同化、脂肪酸合成以及氧化胁迫保护过程,这些生物过程影响了胞内acetyl-CoA和NADPH的水平。淀粉和乳酸盐可能改变胞内代谢物平衡,使得流向acetyl-CoA的碳流提高,NADPH再生能力提高。基于以上结论推测黑曲霉 FB2中伏马菌素的生物合成受到acetyl-CoA调控。
3.2黑曲霉分泌蛋白质组
黑曲霉葡萄糖淀粉酶基因 (glaA) 启动子和glaA信号肽序列广泛用于异源表达载体的构建。然而培养基中的碳源种类对glaA的表达有不同作用。研究发现,木糖强烈抑制glaA的表达,然而麦芽糖是 glaA基因启动子的强诱导剂[21]。Lu等[21]发现木糖或麦芽糖对分泌蛋白质组的组分影响很大,但是对胞内蛋白质组影响较小。另一方面,培养基条件的差异对胞内蛋白质组影响很大。强化表达分子伴侣,低表达折叠酶和高表达空泡蛋白酶使得受控制的反应器比摇瓶更有利于蛋白的生产。
2011年以前,对黑曲霉蛋白分泌途径的研究主要在单一蛋白水平开展,而 Oliveira等[23]应用蛋白质组学的方法来研究高水平分泌条件下黑曲霉分泌蛋白质组和微粒体蛋白质组分(M icrosomal protein composition) 的改变。共识别了102个分泌蛋白和1 126个微粒体酶。以山梨醇作为碳源时,D-麦芽糖和D-木糖的诱导分别提高了胞外淀粉降解酶,木聚糖降解酶和微粒体酶的产量,如D-麦芽糖诱导GlaA,D-木糖诱导 β-木糖酶。在麦芽糖诱导下与翻译、囊泡运输和代谢作用相关的微粒体蛋白表达水平更高,而木糖诱导下,与线粒体相关的微粒体蛋白表达水平更高。在两种诱导条件下高表达的微粒体酶主要作为内质网相关性降解(ER-associated degradation,ERAD) 途径和线粒体蛋白,促进了ATP依赖型蛋白酶体的装配。
Braaksma等[24]通过结合其他相似曲霉的基因模型进行预测,提高了黑曲霉预测的分泌蛋白质组蛋白的数量,避免基因模型不准确造成的预测困难。作为计算机模拟预测的补充,采用鸟枪法蛋白质组学 (Shotgun proteom ics) 分析与生长、碳源消耗相关的分泌蛋白质组。其中,预测的 200个带有信号肽的蛋白被确认是分泌蛋白。对蛋白质组中缺少同源序列的蛋白信号肽预测的准确度达到 85%。最终通过实验确认了计算机模拟的预测结果。
K rijgsheld等[25]将黑曲霉N402菌落划分为5个同心圆区域,采用稳定的二甲基同位素标记处于 5个同心圆区域的分泌蛋白质组,培养条件为以木糖作为碳源、经过或未经过放线菌酮处理。放线菌酮阻断了菌落的边缘蛋白的分泌,但是,相比于未被处理的菌落,经放线菌酮处理过的菌落的中心区域酶分泌能力增强,因为细胞壁被局部降解。在放线菌酮处理过的菌落中共识别到124个蛋白,其中19个分泌蛋白此前从未被识别,中心区域和邻近边缘区域的 35个蛋白的表达量是非添加放线菌酮条件下的 4倍以上,且53个蛋白不存在于非放线菌酮处理条件下的菌落。此外,每个同心区域的分泌组的组分都有差异,说明黑曲霉蛋白分泌具有空间特异性。在之前研究的基础上,Krijgsheld等[26]继续研究,发现孢子形成可抑制蛋白分泌。分别构建了ΔflbA和ΔbrlA (分生孢子形成的中心调节子基因缺失) 的黑曲霉N402突变株。ΔflbA突变株影响了整个菌落的孢子形成和蛋白分泌,但是 ΔbrlA突变株没有改变蛋白的空间分泌。通过定量蛋白质组分析ΔflbA突变株5个同心圆区域的分泌组,共识别到 138个带有信号肽序列的分泌蛋白,18个蛋白从未在黑曲霉的分泌蛋白质组中被报道过,101个蛋白之前没有在野生型黑曲霉N402菌落中被识别。flbA基因失活改变了分泌蛋白在不同空间的分布,并获得了一个更加复杂的分泌蛋白质组。
蛋白糖基化对其在细胞中的生物活性和分拣具有重要的作用,但是目前在大多数真菌中已知的 N端糖基化位点非常有限。Wang等[27]通过应用一个采用酰肼基修饰的磁珠对固相的糖肽进行富集,提出了第一个黑曲霉N端糖基化位点大规模图谱。在黑曲霉的分泌蛋白质组和全细胞裂解液中分析N端糖基化位点,从330 个N端糖蛋白中识别到847个N端糖基化位点。从全细胞裂解液中识别的N糖蛋白主要位于原生质膜、内质网、高尔基体、溶酶体和液泡内,说明N糖蛋白在蛋白分泌途径中的重要作用。此外,序列比对发现,这些糖蛋白还参与很多生物过程,如基因调控、信号转导、蛋白折叠和装配、蛋白修饰和糖代谢途径等。N端糖基化位点的广泛分布,且部分多糖占据了许多蛋白的特殊位点,为从功能上研究蛋白质的N端糖基化作用,及其在黑曲霉中的生物技术应用提供了重要的初始信息。
3.3黑曲霉跨膜蛋白质组
目前,对木质纤维素水解酶以及调控这类酶表达的分子机制已经得到广泛的研究,但是对于黑曲霉中葡萄糖的转运机制研究较少。理解黑曲霉的转运组对提高菌种改造水平和优化过程设计非常重要。Sloothaak等[28]首次研究黑曲霉跨膜转运蛋白质组,采用新开发的隐马尔可夫模型 (Hidden markov model, HMM) 分析pH 3.5时,高浓度和低浓度葡萄糖条件下黑曲霉跨膜相关的蛋白质组。通过结合黑曲霉跨膜蛋白质组的蛋白丰度值与 HMMgluT值,识别到两个新的可能的高亲和力的葡萄糖转运子。并通过酿酒酵母葡萄糖转运子敲除突变株确定了MstG和M stH的功能和生化特性,为研究提高底物利用率提供了新的角度。
4 黑曲霉代谢物组学研究
代谢物组定义为一个生物体或一群细胞在特定时空条件下所有代谢物的集合,这些代谢物是与生物体生理表型相关度最高的一个组学层次。可靠的胞内代谢物组数据的获取和代谢通量分析是揭示细胞真实代谢状态的直接证据,对于提高代谢工程和工业发酵过程优化的理论和实践水平也具有重要意义。
本实验室[29]通过研究黑曲霉产酶突变株GAM 15和模式株CBS 513.88在氧限制的条件下发酵生理参数和代谢流分布的差异,确定了黑曲霉生产糖化酶过程中的一些限制性因素。对副产物的分析发现,突变株的副产物 (草酸和柠檬酸) 积累量远远小于野生株。此外,还发现草酸的形成与菌体的比生长速率具有协同关系,因此进入氧限制条件后,草酸的合成速率降低使得更多的碳流流向糖化酶合成,这也成为氧限制条件下糖化酶产量提高的重要原因。基于本实验室构建的黑曲霉中心碳代谢网络,利用代谢流平衡分析 (Flux balance analysis,FBA) 计算胞内代谢通量分布。结果表明,核糖和还原力是限制菌体生长的主要因素。在氧限制阶段,糖化酶前体氨基酸的供给成为合成糖化酶主要的限制性因素,因此可通过改造氨基酸合成相关的基因提高氨基酸的合成,从而加速糖化酶的合成并提高底物利用效率。
此外,本实验室[30]结合同位素标记和13C代谢流分析方法,研究黑曲霉糖化酶高产菌株DS03043和野生株CBS 513.88的代谢流差异和调控机制。发现黑曲霉DS03043细胞生长速率、葡萄糖利用率和糖化酶产量更高,但是草酸和柠檬酸积累较少。这是因为相比于野生株,高产菌株DS03043中更多的碳流流向氧化磷酸化途径从而降低了TCA循环的碳流,减少了副产物的积累,从而提高了糖化酶的产量。这一结论也与之前的研究结果吻合。而野生菌 CBS 513.88的氧化还原水平较高,NADH的再生和消耗无法平衡,导致了副产物草酸的积累,同时使得草酰乙酸 (OAA) 和磷酸烯醇式丙酮酸(PEP) 在胞内积累,从而降低了葡萄糖的摄入速率,对产物的合成不利。
Wu等[31]发现天蓝色链霉菌和黑曲霉共培养可以显著提高两种菌株抗生素的生产能力。采用基于核磁共振 (Nuclear magnetic resonance,NMR) 的代谢物组学与多元数据结合分析的方法揭示了共培养方式影响黑曲霉和天蓝色链霉菌中次级代谢途径的机理。黑曲霉在共培养方式下可以合成环二肽环 (Phe-Phe)、2-羟基苯乙酸和苯乙酸这 3种在黑曲霉单独培养条件下无法合成的化合物。这是由于黑曲霉苯基丙氨酸代谢途径受到天蓝色链霉菌分泌的物质的诱导,启动了Phe-Phe和苯乙酸合成途径相关基因的表达。因此,微生物共培养与基于NMR的代谢物组学结合的方法可能成为发现新天然产物的有效途径。
5 基因组规模的代谢网络模型
基因组代谢网络模型 (Genome-scale metabolic network model, GSMM) 是由基因-蛋白质-生化反应关联组成的特定的微生物代谢网络,能够帮助更全面地理解细胞的生理特性和代谢能力,并通过整合基因组学、转录组学、蛋白质组学等多组学数据分析细胞的遗传,表观遗传和代谢特性,从全局水平上指导高效、定向调控和优化工业微生物的生产过程和菌种改造[32-33]。高质量的基因组规模代谢网络模型的构建是菌株设计和多层次组学纵向整合的重要基础。大量组学数据的涌现为黑曲霉基因组网络模型的构建和升级提供了必要的基础条件。
基于黑曲霉基因组和文献组数据,帝斯曼首次构建了含有 1 069个反应的黑曲霉 CBS 513.88中心代谢途径的网络模型。通过重新构建的代谢网络解释了黑曲霉高效生产柠檬酸,葡萄糖酸的机制[2]。2007年,孙际宾等对黑曲霉菌株ATCC9029原始的基因组序列进行注释。基于CBS 513.88和ATCC 9029这两株黑曲霉的基因组注释信息,首次构建了黑曲霉全基因规模的代谢网络模型,包括988个特异性酶、2 443个反应和2 349个代谢物[5]。
Nielsen小组[34]2008年发表在Mol Syst Biol的文章在黑曲霉代谢网络模型研究方面具有里程碑式的意义。基于黑曲霉文献组 371篇文献重新构建的黑曲霉 iMA 871代谢网络模型并绘制了代谢网络图包含 1 190个生物化学特异反应,871个开放阅读框,同工酶的存在使得反应数提高到2 240个。相比于之前Sun等[5]自动构建的网络,Nielsen等人工重新构建的网络虽然在规模上有所减小,但是准确性更高。采用文献中的得率、通量分布和转录组的数据对模型的预测结果进行验证,证明了模型的可靠性。
在 2008年黑曲霉基因组网络模型的基础上,本实验室根据最新的黑曲霉基因组注释结果和生理学实验数据对其进行升级。在原有模型的基础上对所有的代谢物进行了重新的注释整理并对化学反应方程式重新配平。重新测定了黑曲霉的详细菌体组成并对能量参数进行重新的拟合。和原有模型比较,新模型中总反应总数从1 380个增加至1 727个,独立代谢物个数由775个增加至868个,独立ORF个数由871个增加至1 138个,反应区间在胞外、胞质和线粒体的基础上增加了过氧化物酶体反应区间。在原有模型基础上我们大幅增加了维生素和辅因子的胞内合成途径,进一步完善了细胞的代谢网络功能。对于升级的基因组网络模型,我们利用恒化实验数据,13C代谢流和代谢物组学数据对模型进行验证,发现新模型的预测值与生理实验数据吻合,表明新模型具有良好的预测性能。利用升级的模型,我们能够预测黑曲霉在大部分碳源的生长情况,同时还预测了菌株生长的必需基因,最后利用该模型我们预测了氧限制对有机酸分泌的影响。上述研究表明,升级的黑曲霉基因组网络模型在规模和应用范围等方面有较好的改进,为黑曲霉的系统代谢工程改造提供了有力的基础[35]。
6 多组学的整合研究与应用
在微生物从反应器提供的环境吸取营养进行繁殖并产生代谢产物的过程中,基因组是稳定的信息源,环境是可控的物质源,两者相互作用后依次产生转录组、蛋白质组、代谢物组和代谢流等组学数据,并以宏观生理参数 (包括菌丝形态、耗糖速率、耗氧速率等) 来体现其群体的生长和生理状态。各组学数据之间有密切关系,也各有其优缺点。比如转录产物存在降解速度快的问题,影响到测序解析的准确性,也有许多转录产物并不翻译为功能性蛋白质;而蛋白质组学中对许多微量蛋白质的分离鉴定还存在技术难度;代谢物组对发酵产物中复杂的小分子化合物的准确定量也未尽如人意等。因此,将多层次组学结合起来,互相参考,有助于提高数据分析的可靠性,对于菌株的全局理性设计和遗传改造有指导意义。
目前黑曲霉中转录组与蛋白质组相结合的研究方法比较常见。黑曲霉分泌系统具有一个良好的蛋白酶质量控制机制,与高效的内质网相关性降解ERAD途径密切相关。而且黑曲霉的分泌系统与哺乳动物和酿酒酵母的分泌系统部分相似[2]。Jacobsa等[32]比较了黑曲霉出发菌株与3株过表达菌株 (分别是异源脂肪酶、同源的蛋白酶和水解酶) 的转录组和蛋白质组,通过相关性分析寻找与酶表达量正相关的、蛋白质组数据和转录组数据变化趋势一致的基因。发现在酶过表达菌株中,蛋白和基因水平一致上调的蛋白主要包括参与碳和氮代谢的蛋白、(氧化) 压力响应蛋白、参与蛋白折叠和 ERAD有关的蛋白。可能意味着在酶高产菌株中,营养代谢、氧化供能、蛋白质合成和折叠加工等途径的活力都有明显上升。其中只有蛋白降解途径的蛋白在所有过表达菌株中都上调表达,说明该途径是进行菌种改造最有潜力的途径。根据这一研究结果,他们通过采用gus (编码β-葡萄糖醛酸酶) 作为报告基因来研究黑曲霉中异源蛋白的生产对内质网膜扩大以及液泡数量增多等方面的影响。在过表达gus的黑曲霉菌株中敲除了ERAD调控因子doaA基因以减少目的蛋白的降解,并过表达寡糖基转移酶sttc基因以增加目的蛋白的糖基化有助于蛋白折叠,结果确实小幅提高了GUS的表达。Carvalho等[33]在原始菌株——含有单拷贝或多拷贝葡萄糖氧化酶葡萄糖醛酸酶 (GlaGus) 融合基因的黑曲霉中构建了ERAD途径部分基因的缺失突变株。敲除多拷贝GlaGus过表达菌株中的derA基因使得细胞内GlaGus蛋白量提高了6倍。以上结果提示,敲除ERAD途径的一些基因,结合异源蛋白的多拷贝表达可促进目的蛋白在胞内的积累,同时降解程度降低,因此不失为在曲霉属宿主中提高异源蛋白表达量的良策。
以各组学数据为基础可以分别构建得到转录调控网络、信号转导网络、分子相互作用网络和代谢途径网络等,它们可以通过基因组代谢网络模型互相联接,因此构建以基因组代谢网络模型为中心,能反映细胞内各组分相互作用关系的全细胞模型未来的发展方向。但由于难度很大,目前全细胞网络模型的建立仍处于初级阶段,仅有少数几个模式菌报道过细胞整合网络模型的构建[36]。
Vongsangnak等[37]通过全基因组代谢网络模型来整合分析转录组数据和代谢流计算结果,确定了米曲霉中参与α-淀粉酶合成和分泌、核苷酸代谢和氨基酸代谢重要的组分 (基因、酶、蛋白和代谢物),可能是提高工业菌株酶产量的潜在靶点。
为分析黑曲霉环境pH和产酸之间的关系,Andersen等[38]将全基因组代谢网络与比较基因组和转录组数据整合,从系统水平分析黑曲霉高效产酸机制。优化了的胞外质子生成的全基因组网络模型,能够再现柠檬酸和草酸合成偏好的pH值。分析pH 2.5、4.5和 6.0这3种条件下的转录组差异并进行聚类分析,共识别了162个基因簇,它们在不同pH条件下具有不同的转录模式。识别到了pH依赖型顺式作用启动子元件以及4种受pH调控的次级代谢产物基因簇。此外,通过比对发现,构巢曲霉 pH调控Pal/PacC途径也存在于黑曲霉中。多组学数据与代谢网络模型的整合分析,为指导黑曲霉高效产酸,研究工业生产过程中pH响应的转录调控机制提供了理论依据。
7 曲霉属基因组数据库
至今为止,已经完成数百个真菌基因组的测序工作,基因组测序数据正在呈井喷式增长。丰富的基因组信息为研究真菌的多样性提供了新方向。然而,进一步解析和理解真菌生活模式这一复杂的生物学机制,需要超越基因组的功能研究。组学技术的发展,能够通过高通量的方法获得大量的真菌基因功能数据 (转录组,蛋白质组等等)。但是由此产生的大量组学数据对数据处理和分析是一种挑战[39]。近年来,随着曲霉菌属公共数据库的建立,能够更加有效地管理和处理海量的基因组数据,发现曲霉菌属序列间的科学关联,为曲霉菌属比较基因组学的研究奠定了基础。
CADRE数据库 (http://www.cadre-genomes. org.uk) 建立于 2001年,是一个用于储存和分析曲霉菌属基因组数据公共的数据库。它可以保存完整的烟曲霉注释后的基因组序列,并提供搜索,分析和可视化真菌基因组特征的工具。CADRE以Ensembl数据库为基础,用来管理测序信息,进行辅助注释并且通过合并构巢曲霉和其他曲霉菌种的基因组数据,促进之后的比较组学研究[40]。自从 2004年首次发布后,CADRE数据库已经扩展到包含8种其他曲霉的注释序列,为国际曲霉研究组提供更多必要的支持。Gilsenan等升级了数据库和软件集,不仅可以更好的管理更加复杂的数据集,通过提供基因组比较数据和高通量数据还可以改进注释信息[41]。
曲霉属基因组数据库 (AspGD;http://www.aspgd.org) 是一个免费的在线曲霉菌属数据库,也是整个真菌研究机构重要的信息来源。AspGD除了作为一个数据库以及访问基因组、转录组和多态性数据的中心,还拥有全面的比较基因组工具,在20个现有的曲霉属基因组中探索预先计算的直系同源基因。AspGD管理员基于构巢曲霉、米曲霉、烟曲霉和黑曲霉这 4种关键曲霉菌种的文献信息完成了基因注释。通过分析公共的转录组数据,表达序列标签,管理员已经反复提高了曲霉菌基因组的结构注释信息。基于最近的RNA-Seq数据,报道了构巢曲霉、米曲霉和烟曲霉基因组结构注释信息重大的改进,更新了超过2 600个位点的信息[42-43]。
Park等[44]采用标准化的流程对真菌转录因子 (Transcription Factors, TFs) 注释,于 2008年构建了真菌转录因子数据库 (FTFD)(http://ftfd.snu.ac.kr)。数据库从 61个真菌和 3个卵菌全基因组数据中预测了75 131个潜在的真菌TFs分别属于61个家族,数据库还能够比较分析 TFs在相同菌种和不同菌种之间的分布以及结构差异,构建所有 TFs的系统进化树。数据库包含 7种曲霉菌种,其中黑曲霉 ATCC 1015和CBS 513.88分别识别到633和708个转录因子。转录因子和转录因子结合位点(Transcription Factor Binding Site, TFBS) 是转录水平调控基因表达的重要组成部分,为深入研究转录因子和转录因子结合位点的作用机制,构建转录调控网络,需要收集、整理已发现的多种TFs和TFBS信息,形成转录因子数据库[45]。转录因子数据库的构建为系统分析真菌转录因子,研究从转录水平调控基因表达起到重要的推动作用。
8 讨论与展望
细胞的生命活动是多层次网络相互调节作用的结果,单一组学的数据不足以全面、真实地反映细胞的代谢活动。基于基因组信息构建的全基因组代谢网络模型在很大程度上只是提供了细胞代谢活动的可能性,具有很大的不确定性。相比于基因组学,转录组学的研究更具有时间和空间特异性。细胞在不同的生长阶段,特定的条件下,转录组都具有一定差异。通过新一代RNA-Seq技术能够提供基因结构,基因功能,新转录本,可变剪切现象等更多的信息。分析基因的差异表达能够帮助研究者深入理解环境和生物过程对基因表达的重要影响。分析黑曲霉在蛋白分泌表达、能量代谢、信号转导、营养物利用和生长生育等方面的差异,为调控产物的合成途径,为黑曲霉工业生产菌株的改造提供靶点,为过程调控提供分子机制的依据。但是转录组学目前还存在取样点合理性、RNA降解快难以提取高质量RNA和无高精度参考基因组条件下转录组分析结果准确性等问题,为转录组学的发展提出了挑战。转录组学也搭建起蛋白质组学与基因组学之间的桥梁,因为单一的蛋白质组学不足以清楚地鉴定基因功能,需要转录组的数据加以印证。转录组学和蛋白质组学虽然一定程度上能间接反映菌体状态的变化,但由于酶的活力受变构反馈控制以及胞内代谢物浓度的影响,mRNA和蛋白质丰度往往无法与胞内代谢通量直接关联。代谢物组学技术采用高通量和高灵敏度的分析手段,基因和蛋白表达极小的变化都能在代谢产物上得到放大,所以代谢物组学是理解生理过程的有效途径,它能真正地反映菌株的表型,可以用来弥补基因组学、转录组学和蛋白质组学的不足[17]。胞内代谢通量和胞内代谢物池大小的变化提供了细胞代谢活动的直接证据且与目标产物的合成密切相关,因此开展以13C代谢流和代谢物组学为核心的纵向组学整合研究十分必要。研究表明,仅仅基于基因注释构建的全基因组代谢网络模型不足以准确地反映细胞代谢过程,精确预测菌体表型,因此从系统生物学的角度将多层次组学整合起来,构建全细胞网络模型以从全局高度指导细胞工厂的理性设计和发酵过程优化是将来极具吸引力的发展方向,但数据整合和模型化的数学方法仍需探索。比如转录调控网络与 GSMM 的整合上已有GeneForce, PROM 和 Regulatory FBA (rFBA) 3种算法,integrative om ics-metabolic analysis (IOMA) 算法用于分析蛋白质-代谢物组整合的基因组代谢网络[46]。
另外,目前的黑曲霉的组学研究较多集中在碳源种类和水平的影响,以及分泌途径的组分和调控等,而发酵过程中关键生理参数变化时期的组学响应还缺乏系统深入的研究。例如,黑曲霉在液体发酵过程中呈现出多变的菌体形态[47],主要分为分散菌丝、成簇、成团结球 3类[48]。丝状真菌的形态变化带来的粘度变化直接影响发酵过程氧和营养的传递过程,进一步诱导了菌丝形态分化,并且与产物的大量合成也密切相关。因此,研究黑曲霉发酵过程中生理参数 (比生长速率、呼吸熵、糖耗速率和菌丝形态等等) 转变时期的多组学响应,揭示过程调控手段对菌体代谢和产物合成的作用机制,对优化黑曲霉工业生产过程调控和菌株遗传改造也具有重要意义。
REFERENCES
[1] Baker SE. Aspergillus niger genom ics: past,present and into the future. M ed M ycol, 2006,44(Suppl 1): 17-21.
[2] Pel HJ, de W inde JH, A rcher DB, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88, Nat Biotechnol, 2007, 25(2): 221-231.
[3] Yin C, Wang B, He P, et al. Genom ic analysis of the aconidial and high-performance protein producer, industrially relevant Aspergillus niger SH2 strain. Gene, 2014, 541(2): 107-114.
[4] Kwon M J, Jorgensen TR, Nitsche BM, et al. The transcriptom ic fingerprint of glucoamylase over-expression in Aspergillus niger. BMC Genom ics, 2012, 13: 701.
[5] Sun JB, Lu X, Ursula R, et al. Metabolic peculiarities of Aspergillus niger disclosed by comparative metabolic genom ics. Genome Biol,2007, 8(9): R182.
密切观察患者各生命体征,所有患者实施CT增强扫描过程中护理人员需观察其一次穿刺率与造影剂外渗率,其中前者评价标准为对患者穿刺后,观察到回血后注入药液,注射液无外渗则说明穿刺成功;反之穿刺失败;后者重要评价标准为完成穿刺后注射液无泄漏情况,但增强扫描时造影剂有外渗情况产生,导致患者存在局部疼痛感。
[6] Andersen MR, Nielsen J. Current status of systems biology in Aspergilli. Fungal Genet Biol,2009, 46(Suppl 1): 180-190.
[7] Shi SB, Chen T, Zhao XM. Transcriptome platforms and application to metabolic engineering, Chin J Biotech, 2010, 26(9): 1187-1198 (in Chinese).
史硕博, 陈涛, 赵学明. 转录组平台技术及其在代谢工程中的应用. 生物工程学报, 2010, 26(9): 1187-1198.
[8] Andersen, MR, Salazar, MP, Schaap, PJ. Comparative genom ics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res,2011, 21(6): 885-897.
[9] Jørgensen TR, Nitsche BM, Lamers GE, et al. Transcriptom ic Insights into the physiology of Aspergillus niger approaching a specific grow th rate of zero. Appl Environ M icrob. 2010, 76(16):5344-5355.
[10] JØrgensen TR, Goosen T, van den Hondel CA, et al. Transcriptom ic comparison of Aspergillus niger grow ing on two different sugars reveals coordinated regulation of the secretory pathway. BMC Genom ics, 2009, 10(1): 44.
[11] Yuan XL, van der Kaaij RM, van den Hondel CAM JJ, et al. Aspergillus niger genome-w ide analysis reveals a large number of novel alpha-glucan acting enzymes w ith unexpected expression profiles. M ol Genet Genom ics, 2008,279(6): 545-561.
[12] Vongsangnak W, Salazar M, Hansen K, et al. Genome-w ide analysis of maltose utilization and regulation in aspergilli. M icrobiology, 2009,155(12): 3893-3902.
[14] de Souza WR, de Gouvea PF, Savoldi M, et al. Transcriptome analysis of Aspergillus niger grown on sugarcane bagasse. Biotechnol Biofuels,2011, 4(1): 40.
[15] Pullan ST, Daly P, Delmas S, et al. RNA-sequencing reveals the complexities of the transcriptional response to lignocellulosic biofuel substrates in Aspergillus niger. Fungal Biol Biotechnol, 2014, 1(1): 3.
[16] van Munster JM, Dalya P, Delmas S, et al. The role of carbon starvation in the induction of enzymes that degrade plant-derived carbohydrates in Aspergillus niger. Fungal Genet Biol, 2014, 72: 34-47.
[17] Nitsche BM, JØrgensen TR, Akeroyd M, et al. The carbon starvation response of Aspergillus niger during submerged cultivation: insights from the transcriptome and secretome, BMC Genom ics,2012, 13: 380.
[18] de Souzaa WR, Maitan-A lfenasb GP, de Gouvêa PF, et al. The influence of Aspergillus niger transcription factors A raR and X lnR in the gene expression during grow th in D-xylose,L-arabinose and steam-exploded sugarcane bagasse. Fungal Genet Biol, 2013, 60: 29-45.
[19] Novodvorska M, Hayer K, Pullan ST, et al. Transcriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genom ics, 2013, 14(1): 246.
[20] van Leeuwen MR, K rijgsheld P, W yatt TT, et al. The effect of natamycin on the transcriptome of conidia of Aspergillus niger. Stud M ycol, 2012,74(1): 71-85.
[21] Lu X, Sun JB, Nim tz M, et al. The intra- and extracellular proteome of Aspergillus niger grow ing on defined medium w ith xylose or maltose as carbon substrate. M icrob Cell Fact,2010, 9(17): 23.
[22] SØrensen LM, Lametsch R, Andersen MR, et al. Proteome analysis of Aspergillus niger: lactate added instarch-containing medium can increase production of themycotoxin fumonisin B2by modifying acetyl-CoA metabolism. BMC M icrobiol, 2009, 9: 255-274.
[23] de Oliveira JMPF, van Passel MW J, Schaap P, et al. Proteom ic analysis of the secretory response of Aspergillus niger to D-maltose and D-xylose. PLoS ONE, 2011, 6(6): e20865.
[24] Braaksma M, Martens-Uzunova ES, Punt PJ, et al. An inventory of the Aspergillus niger secretomeby combining in silico predictions w ith shotgun proteom ics data. BMC Genom ics, 2010, 11(1): 584.
[25] K rijgsheld P, A ltelaar AFM, Post H, et al. Spatially resolving the secretome w ithin the mycelium of the cell factory Aspergillus niger. J Proteome Res, 2012, 11(5): 2807-2818.
[26] Krijgsheld P, Nitsche BM, Post H, et al. Deletion of flbA results in increased secretome complexity and reduced secretion heterogeneity in colonies of Aspergillus niger. J Proteome Res, 2013, 12(4): 1808-1819.
[27] W ang L, A ryal UK, Dai ZY, et al. M apping N-linked glycosylationsites in the secretome and whole cells of Aspergillus niger using hydrazide chem istryand mass spectrometry. J Proteome Res,2012, 11: 143-156.
[28] Sloothaak J, Odoni DI, de Graaff LH, et al. Aspergillus niger membrane-associated proteome analysis for the identification of glucose transporters. Biotechnol Biofuels, 2015, 8: 150.
[29] Chen XF, Lu HZ, Tang W J et al. Comparison of two Aspergillus niger mutant and w ild strains based on q-rate and flux balance analysis. J Chin Biotechnol, 2014, 34(8): 35-40 (in Chinese).
陈香粉, 鲁洪中, 唐文俊. 基于比速率及代谢流的黑曲霉突变株和野生株分析, 中国生物工程杂志, 2014, 34(8): 35-40.
[30] Lu HZ, Liu XY, Huang MZ, et al. Integrated isotope-assisted metabolom ics and13C metabolic flux analysis reveals metabolic flux redistribution for high glucoamylase production by Aspergillus niger. M icrob Cell Fact, 2015, 14(1): 147.
[31] W u CH, Zacchetti1 B, Ram AFJ, et al. Expanding the chem ical space for natural products by Aspergillus-Streptomyces co-cultivation and biotransformation. Sci Rep, 2015, 5: 10868.
[32] Jacobsa DI, Olsthoorna MMA, Maillet I, et al. Effective lead selection for improved protein production in Aspergillus niger based on integrated genom ics. Fungal Genet Biol, 2009,46(Suppl 1): S141-S152.
[33] Carvalho NDSP, A rentshorst M, Kooistra R, et al. Effects of a defective ERAD pathway on grow th and heterologous protein production in Aspergillus niger. Appl M icrobiol Biotechnol,2011, 89(2): 357-373.
[34] Andersen MR, Nielsen ML, Nielsen J. Metabolic model integration of the bibliome, genome,metabolome and reactome of Aspergillus niger. M ol Syst Biol, 2008, 4: 178.
[35] Lu HZ, Ouyang LM, Xia JY, et al. A comprehensive reconstruction and verification of Aspergillus niger genome-scale metabolic network model. ME Summ it 2015 Abstracts,2015: 120.
[36] Xu ZX, Zheng P, Sun JB. Reconstruction of whole cell network and design of cell factory. Prog Biochem Biophys, 2014, 41(2): 105-114(in Chinese).
徐自祥, 郑平, 孙际宾. 全细胞网络重建与细胞工厂设计. 生物化学与生物物理进展, 2014,41(2): 105-114.
[37] Vongsangnak W, Hansen K, Nielsen J. Integrated analysis of the global transcriptional response to α-Amylase over-production by Aspergillus oryzae. Biotechnol Bioeng, 2011, 108(5): 1130-1139.
[38] Andersen MR, Linda L, Nielsen J, et al. System ic analysis of the response of Aspergillus niger to ambient pH. Genome Biol, 2009, 10(5): R47.
[39] Aguilar-Pontes MV, de Vries R, Zhou MM. (Post-) Genom ics approaches in fungal research. Brief Func Genom ics, 2014, 13(6): 424-439.
[40] Mabey JE, Anderson M J, Giles PF, et al. CADRE: the central Aspergillus data REpository. Nucleic Acids Res, 2004, 32(Database issue): D401-D405.
[41] Gilsenan JM, Cooley J, Bow yer P. CADRE: the central Aspergillus data REpository 2012. Nucleic Acids Res, 2011, 12: 1-7.
[42] Arnaud MB, Cerqueira GC, Inglis DO, et al. The Aspergillus genome database (AspGD): recent developments in comprehensive multispecies curation, comparative genom ics and community resources. Nucleic Acids Res, 2012, 40(Databaseissue): D653-D659.
[43] Cerqueira GC, Arnaud MB, Inglis DO, et al. The Aspergillus genome database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res, 2014, 42(Database issue): D705-D710.
[44] Park J, Park J, Jang S, et al. FTFD: an informatics pipeline supporting phylogenom ic analysis of fungal transcription factors. Bioinformatics, 2008,24(7): 1024-1025.
[45] Chen HF, Wang JK. The databases of transcription factors. Hereditas, 2010, 32(10): 1009-1017 (in Chinese).
陈鸿飞, 王进科. 转录因子相关数据库. 遗传,2010, 32(10): 1009-1017.
[46] Liu LM, Liu T, Zou W. Constraint-based algorithms for genome scale metabolic model-a review. Chin J Bioproc Eng, 2012, 10(6): 70-77(in Chinese).
刘立明, 刘婷, 邹伟. 基因组规模代谢网络模型的约束算法及其应用. 生物加工过程, 2012,10(6): 70-77.
[47] Tang W J, Xia JY, Chu J, et al. Development and application of morphological analysis method in Aspergillus niger fermentation. Chin J Biotech,2015, 31(2): 291-299 (in Chinese).
唐文俊, 夏建业, 储炬, 等. 黑曲霉发酵过程中菌体形态的分析方法建立及应用. 生物工程学报, 2015, 31(2): 291-299.
[48] Metz B, Kossen NWF. The grow th of molds in the form of pellets-a literature review. Biotechnol Bioeng, 1977, 19(6): 781-799.
(本文责编 陈宏宇)
Progress in omics research of Aspergillus niger
Yufei Sui*, Liming Ouyang*, Hongzhong Lu, Yingping Zhuang, and Siliang Zhang
State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China
Aspergillus niger, as an important industrial fermentation strain, is widely applied in the production of organic acids and industrial enzymes. With the development of diverse omics technologies, the data of genome, transcriptome,proteome and metabolome of A. niger are increasing continuously, which declared the coming era of big data for the research in fermentation process of A. niger. The data analysis from single omics and the comparison of multi-omics, to the integrations of multi-omics based on the genome-scale metabolic network model largely extends the intensive and systematic understanding of the efficient production mechanism of A. niger. It also provides possibilities for the reasonable global optimization of strain performance by genetic modification and process regulation. We reviewed and summarizedprogress in om ics research of A. niger, and proposed the development direction of om ics research on this cell factory.
December 3, 2015; Accepted: February 17, 2016
Yingping Zhuang. Tel: +86-21-64251257; E-mail: ypzhuang@ecust.edu.cn
Aspergillus niger, genome, transcriptome, proteome, metabolome, om ics integration, genome-scale metabolic network model
Supported by: Open Funding Project of the State Key Laboratory of Bioreactor Engineering.
*These authors contributed equally to this study.
生物反应器工程国家重点实验室开放课题资助。
网络出版时间:2016-03-07 网络出版地址:http://www.cnki.net/kcms/detail/11.1998.Q.20160307.0956.001.html
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
中国土壤与肥料(2021年5期)2021-12-02
昆钢科技(2021年6期)2021-03-09
国际口腔医学杂志(2019年3期)2019-05-31
天然产物研究与开发(2018年2期)2018-04-04
中国酿造(2016年12期)2016-03-01
中国酿造(2016年12期)2016-03-01
电源技术(2016年9期)2016-02-27
大连工业大学学报(2015年4期)2015-12-11
医学研究杂志(2015年11期)2015-06-10
