
烷基化果胶的微波合成及其理化性质研究
2016-09-12 01:05:12俸思洁王玲华梁瑞红
食品工业科技 2016年7期
俸思洁,王玲华,梁瑞红,陈 军,刘 伟
(南昌大学 食品科学与技术国家重点实验室,江西南昌 330047)
烷基化果胶的微波合成及其理化性质研究
俸思洁,王玲华,梁瑞红*,陈军,刘伟
(南昌大学 食品科学与技术国家重点实验室,江西南昌 330047)
为考察微波辅助果胶烷基化的可行性,本文研究了果胶与溴代己烷的比例、微波功率以及辐射时间对烷基化果胶取代度的影响,以及所得烷基化果胶的理化性质。结果表明:提高果胶与溴代己烷的比例、增加微波功率及辐射时间均有助于取代度的提高,但升高到一定程度后,均趋向平稳。在功率为320 W,果胶:溴代己烷摩尔质量比为1∶1,反应时间为240 s时,取代度为4.81%,优于传统方法用时24 h所得衍生物的取代度(3.60%)。这说明微波辐射能有效降低果胶与溴代己烷的反应时间,提高烷基化取代度。且随着取代度增大,微波辅助烷基化果胶比原果胶具有更高的表观粘度、乳化性(EC)和乳化稳定性(ES)。微波辅助烷基化果胶最大EC和ES能达57.29%和93.61%,优于传统方法制备的烷基化果胶和原果胶。
烷基化果胶,合成,微波辐射,表观粘度,乳化性
果胶是一类由D-半乳糖醛酸残基以α-1,4糖苷键连接组成的植物细胞壁复杂多糖,主要以原果胶、果胶和果胶酸等形态存在于植物体的细胞壁和细胞间层中[1];也是一种被公认的安全无毒且使用无任何限制的高分子多糖,常作为凝胶剂、增稠剂、组织改良剂、乳化剂和稳定剂等广泛应用于食品、医药和化妆品等行业。近些年,人们试图对果胶的一些结构进行人为的修饰,来增强或者改变其功能性质,以满足市场对创新和功能性产品的需求。因此,对果胶进行修饰的研究报道也越来越多。果胶自身的分子结构决定了化学修饰方法的多样性,主要包括酰胺化[2-3]、季铵化[4-5]、硫酸化[6-8]和烷基化,其中果胶的烷基化即在果胶的C-6羧基基团或C-2、C-3羟基基团上引入烷基链,主要分为羧酸的烷基化以及羟基的烷基化。
本实验室前期采用烷基化法制得的果胶,虽然其表观粘度、乳化性及稳定性有了很大的提高,但烷基化时间长达24 h[9]。自1986年首次报道微波合成技术以来[10-11],因其在温和的反应条件下,能快速提高反应速率、提高产物纯度和产率,且操作方便[12],而被广泛地应用于有机合成领域。但微波用于果胶烷基化修饰的研究尚未见文献报道。本实验通过对微波辅助果胶烷基化反应的可行性、烷基化果胶产物结构与性质的变化研究,为扩大果胶及其衍生物的应用提供理论依据。
1 材料与仪器
1.1材料与试剂
柑橘果胶Sigma Aldrich公司;溴代己烷、己醇、十二烷、环戊烷和四丁基氢氧化铵(TBA+OH-)(25 wt%水溶液)阿拉丁试剂(上海)有限公司;无水乙醇、无水丙酮、氯化钠、叠氮钠、氢氧化钠、二甲基亚砜DMSO等试剂均为国产分析纯。
Agilent 6890型气相色谱仪美国安捷伦公司;TDL-5-A型台式离心机上海安亭科学仪器厂;Nicolet 5700型傅里叶红外光谱仪美国Thermo公司;Avance Ⅲ 600型核磁共振谱仪德国Brucker公司;Discovery DHR-2型流变仪美国TA公司;EX1103ZH型电子分析天平北京西杰天平仪器有限公司;DW-86L390型超低温冰箱青岛澳柯玛超低温冷冻设备有限公司;FreeZone®2.5 L型真空冷冻干燥机美国Labconco公司;SHB-3型循环水多用真空泵郑州杜甫仪器厂;X85-2S型恒温磁力搅拌器上海梅颖浦仪器仪表制造有限公司;SD101-0A型电热恒温鼓风干燥箱南通金石实验仪器有限公司;V-1001型旋转蒸发仪上海爱朗仪器厂;XW-80A型旋涡混合器上海精科实业有限公司;Ultra-Turrax T25 basic型分散机德国IKA公司。
1.2实验方法
1.2.1烷基化果胶的合成
1.2.1.1微波合成参照文献[13]进行果胶烷基化。具体方法如下:配制2%(w/v)的果胶溶液,在通风橱中加入四丁基氢氧化铵(TBA+OH-)中和溶液pH至7。冷冻干燥后,称取一定量的TBA-果胶粉末溶于二甲基亚砜(DMSO)中,配成2%的溶液。再加入一定量的溴代己烷,混合搅拌均匀后,样品转移到微波炉中,控制一定的微波条件进行反应。反应结束后放置在蒸馏水(6 h更换一次)中透析7 d以除去DMSO,旋转蒸发后获得的浓缩溶液用3倍体积70%的乙醇溶液(含1 mg/mL氯化钠)沉淀出烷基化产物,在4℃下静置24 h,确保果胶分子上的TBA+被过量的Na+取代。过滤后,先用75%的乙醇清洗,然后再用无水乙醇清洗,用氢氧化银验证至没有氯离子,常温常压下干燥,即得到微波辅助烷基化果胶产物。
1.2.1.2常温合成固定果胶(以半乳糖醛酸计算)与溴代己烷的摩尔质量比为1∶1,在室温下搅拌反应24 h。其它条件和方法同1.3.1.1项。
1.2.2影响产物取代度(DS)的因素分析
1.2.2.1微波辐射功率对产物取代度的影响固定果胶(以半乳糖醛酸计算)与溴代己烷的摩尔质量比为1∶0.5,分别在微波辐射功率160、240、320、400、480和560 W下反应90 s,并分别测定产物的取代度,研究微波辐射功率对烷基化果胶取代度的影响,选择最优的辐射功率进行后续实验。
1.2.2.2微波辐射时间对产物取代度的影响分别选取30、60、90、120、180、240 s的辐射时间,果胶(以半乳糖醛酸计算)与溴代己烷的摩尔质量比为1∶0.5和1∶1,在最优的辐射功率下进行反应,并测定产物的取代度,研究果胶与溴代己烷的比例及微波辐射时间对产物取代度的影响。
1.2.3取代度的测定用气相色谱法测定衍生物的取代度[14],色谱条件:色谱柱HP-5;载气(氮气)流速2.5 mL/min;检测器:FID;进样温度为280℃;检测器温度为280℃;程序升温以10℃/min从60升温至200℃,在60、90、150℃保持1 min;进样量2 μL;分流比为1∶10。
1.2.3.1标准曲线的绘制内标液的配制:称取50 mg内标物十二烷于烧杯中,用环戊烷溶解,配成1 mg/mL的内标液。
标准液的配制:一定量的己醇与甲醇溶解定容,得到浓度为2 mg/mL的标准液。用移液管分别取己醇标准液1、2、3、4、5、6 mL于容量瓶中,用甲醇定容至10 mL。吸取1 mL标液和2 mL内标液混合均匀后进样。以标准样品的峰面积/内标物的峰面积为纵坐标,标准样品的质量为横坐标,绘制己醇的标准曲线,线性回归。
1.2.3.2样品取代度的测定100 mg的烷基化果胶加水搅拌至溶解,加入0.4 mol/L的NaOH反应4 h。加入内标液,持续剧烈搅拌,4800 r/min离心15 min后,溶液分层,取上层有机相过滤膜进样,与标准曲线对照得到对应的己醇量。使用WPC6Sx来命名微波烷基化产物,PC6Sx来命名烷基化产物。其中W代表微波辐射,P代表果胶,C6代表共价连接的己烷,x代表取代度DS值。Pctrl则表示不加溴代烷其它步骤与获得烷基化产物相同的对照组。
1.2.4产物分析
1.2.4.1FT-IR分析将一定量的干燥果胶样品或烷基化果胶样品与一定量的KBr粉末混合在玛瑙研磨器中研磨至粉状,在频率范围为4000~400 cm-1下进行红外光谱扫描[15]。收集数据并绘制透射比(%)与波数的函数关系图,用Ominic7.2软件进行数据分析。
1.2.4.21H-NMR分析称取10 mg的原果胶或烷基化果胶样品溶解于0.5 mL的D2O中,在25℃条件下进行1H-NMR检测[16]。
1.2.4.3表观粘度的测定果胶溶于0.025 mol/L的氯化钠溶液中,配制20 g/L的溶液,在室温下搅拌18 h,3000 r/min下离心15 min以去除气泡,测定前4℃冰箱中放置过夜[17]。采用Discovery DHR-2流变仪,温度为(25±1)℃,在剪切速率0.001~10 s-1范围内由低到高进行扫描,得到粘度随剪切速率的变化曲线。
1.2.4.4乳化性分析乳化性质的测定参照文献方法[18]。将果胶及其衍生物样品溶解在蒸馏水中,配成浓度为0.5%(w/w)的溶液,加入0.02%的NaN3作为防腐剂,在溶液中缓缓加入植物油,使得油的质量分数为40%(w/w)。混合液用Ultra-Turrax T25 basic分散机的最大转速分散2 min,制备的乳液倒入15 mL的离心管中,在3000 r/min下离心5 min。测定乳液的总体积Wv和乳化层体积ELvi,乳化能力(EC)计算如下:
EC(%)=(ELvi/Wv)×100
式(1)
上述离心之后的乳液在室温下静置7 d,继续上述实验操作,离心后得到最终的乳化层体积ELvf,乳化稳定性(ES)计算如下:
ES(%)=(ELvf/ELvi)×100
式(2)
1.2.5数据分析本文中所有实验均重复3次,采用Origin 8.0和SPSS16.0对实验数据进行统计分析,样品平均值之间的差异性通过Duncan法比较(p<0.05)。
2 结果与分析
2.1微波辐射条件对取代度的影响
从图1可看出,随着微波功率的增加,烷基化果胶取代度显现先升高后趋向平稳的趋势。在功率为160 W时取代度仅为0.66%,这可能是因为功率太低,微波不能提供足够的能量进行反应[19];而随后随着功率的上升,微波提供的能量不断增大,产物取代度随之急剧上升,当提供的能量突破反应所需的活化能后,反应趋于平缓,在320 W时达到平稳值,此时的取代度为2.90%。
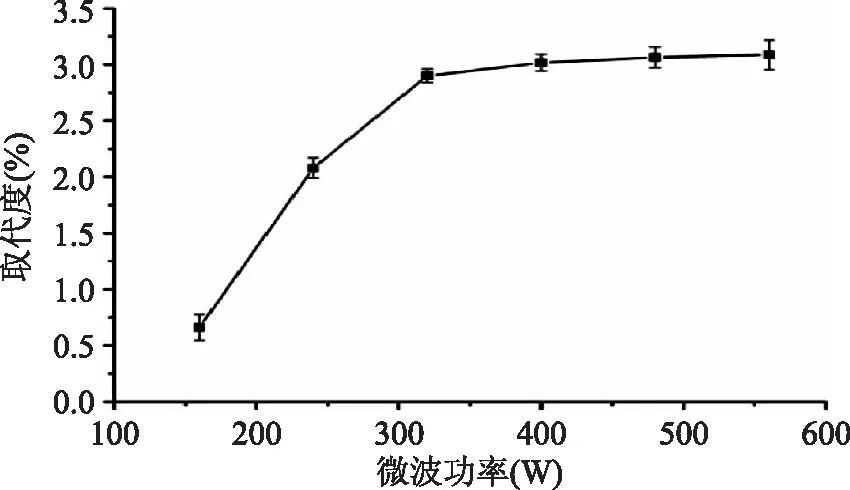
图1 微波辐射功率对取代度DS的影响Fig.1 Effect of mircowave power on the degree of substitution
图2显示,在任意辐射时间下,修饰物DS(果胶∶溴代己烷=1∶1)>DS(果胶∶溴代己烷=1∶0.5),可能是随着溴代己烷使用量的增加,果胶与溴代己烷的接触机会增大,反应越充分。随着微波反应时间的延长,产物取代度不断增大,而后增长趋势变得平缓。对于果胶∶溴代己烷为1∶1的样品,反应时间为30 s时DS就已经达到了2.96%,反应240 s时DS为4.81%,而传统方法获得的衍生物DS为3.60%,用时24 h。因此,微波辅助果胶烷基化在短时间内就能获得高DS。本实验选取DS为2.96%、3.85%以及4.81%的反应物进行后续实验,分别命名为WPC6S2.96、WPC6S3.85和WPC6S4.81。

图2 果胶与溴代己烷的比例以及微波辐射时间对产物取代度的影响Fig.2 Effect of mass ratio of pectin to bromohexane and reaction time on DS
2.2微波辅助烷基化果胶的结构表征
采用傅里叶红外光谱对微波辅助烷基化果胶的化学结构进行分析,并与原果胶以及传统方法获得的烷基化果胶进行比较,其结果如图3所示。果胶的特征吸收峰有:3450.8 cm-1(υ(OH))、2925.9 cm-1(υ(CH))、1744.6 cm-1(υ(C=O)COOMe和υ(COOH))、1620.4 cm-1(υas(COO-))、1150.1 cm-1(υ(COC))和1103.8 cm-1(υ(C-C))。其中,1744 cm-1吸收峰面积与1744、1620 cm-1处总吸收峰面积的比值常用来表示果胶的酯化度。从图3中可以看出,三个微波辅助烷基化果胶样品的这一比值随着取代度的增加而增大,增长趋势与传统方法获得的不同取代度衍生物趋势相似,这表明在微波辅助下,果胶的羧酸基团与溴代己烷发生了酯化反应。且WPC6S3.85与传统方法获得的PC6S3.60红外结构相似,表明微波辐射没有破坏烷基化果胶的结构。

图3 果胶及其微波辅助衍生物的红外光谱图Fig.3 FT-IR spectra of pectin and its microwave-assisted derivatives注:A:原果胶;B:Pctrl;C:WPC6S2.96;D:PC6S3.60; E:WPC6S3.85;F:WPC6S4.81。
由图4可看出,1H-NMR主要由甲基和亚甲基质子信号峰表征,其中1.6 ppm为靠近羰基的亚甲基吸收峰,1.4~1.1 ppm范围内为其余亚甲基的质子信号峰,0.8 ppm为烷基碳链中甲基的吸收峰。从图中我们可以得知,原果胶1H-NMR图谱中无1.6 ppm,1.4~1.1 ppm和0.8 ppm信号峰,这些信号峰在微波辅助烷基化果胶的1H-NMR图谱中才出现,且两处的吸收峰强度随着取代度增多而增强,有力地证明了微波辅助技术在烷基化果胶制备中的成功应用。

表1 果胶及其微波辅助衍生物的乳化能力(EC)和乳化稳定性(ES)
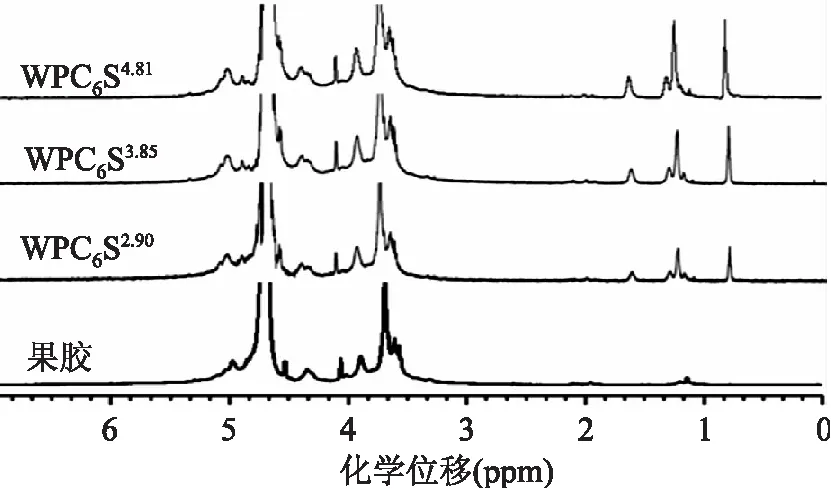
图4 果胶及其微波辅助衍生物的1H-NMR图谱Fig.4 1.D proton NMR spectra of pectin and its microwave-assisted derivatives
注:a实验数据采用均值±标准偏差,*同一行数据,不同字母表示显著性差异(Tukey test,p<0.05)。
2.3微波辅助烷基化果胶的表观粘度
图5为20 g/L果胶溶液25℃时的表观粘度(η)与剪切速率(r)的对数关系图。从图中可以看出,原果胶的表观粘度随着剪切速率的增大而降低,溶液表现出典型的剪切变稀现象。然而三种微波辅助烷基化果胶在低剪切速率下,溶液的表观粘度随着剪切速率的增大先增加,表现出剪切稠化特性;随后粘度随剪切速率增大而降低。这可能是因为剪切速率的变化使得烷基化果胶分子内与分子间的疏水缔合结构相互转变引起的[20]。WPC6S2.96、WPC6S3.85和WPC6S4.81样品在一定剪切速率下分子链扩张伸展,分子内的缔合作用转变成分子间的缔合作用,溶液粘度增加;当剪切速率继续增加时,分子间的疏水缔合结构遭到剪切力破坏,表现为溶液粘度下降。在任意剪切速率下,微波辅助烷基化果胶的表观粘度要大于原果胶的表观粘度,且衍生物的粘度值随着取代度的增大而增加。这种增大的幅度在低剪切速率下较明显。当剪切速率较大时,烷基化果胶的分子间疏水缔合能力遭到破坏导致分子链伸展,溶液粘度降低,表观粘度差异性变小。

图5 果胶及其微波辅助衍生物的表观粘度Fig.5 Apparent viscosity of pectin and its microwave-assist derivatives
2.4微波辅助烷基化果胶的乳化性质
为了验证微波辅助和传统方法获得的烷基化果胶性质方面是否相同,实验还进行了微波辅助烷基化果胶的乳化性质的测定。分别用0.5%(w/w)的果胶及其衍生物溶液制备乳液,乳液离心之后分为三层,少量的油相分布在上层,底层是分散有烷基化果胶的水相,处于两者之间的是乳化层。根据乳化层的体积计算出EC值和ES值,结果如表1所示,WPC6S2.96、WPC6S3.85和WPC6S4.81的乳化能力分别为49.93%、54.17%和57.29%,显著大于未修饰果胶的EC(35.00%)。而在相似的取代度下(如PC6S3.60和WPC6S3.85),微波辅助烷基化果胶与传统烷基化果胶具有相似的乳化能力和乳化稳定性,表明微波能够缩短反应时间,但不影响所得产物的功能性质。但通过微波辅助修饰,能得到比传统方法取代度更好的产物WPC6S4.81,其具有最大的EC(57.29%)和ES(93.61%)。故微波辅助可以提高烷基化果胶的取代度,进而提高其乳化性质。
3 结论
本文研究了微波辐射功率以及辐射时间对烷基化果胶取代度的影响规律。结果表明,微波辐射技术可成功地应用于烷基化果胶的制备,显著缩短反应时间,提高烷基化取代度;产物取代度随功率增大急剧上升,后趋于平缓,在功率为320 W时达到平稳值;反应时间仅240 s时取代度为4.81%,明显大于传统方法反应24 h时的取代度(3.60%);在相似取代度下,微波辅助烷基化果胶与传统方法制备的果胶表现出相似的功能性质。
[1]陈熠,熊远福,文祝友,等.果胶提取技术研究进展[J].中国食品添加剂,2009(3):80-84.
[3]Kratchanova M,Slavov A,Kratchanov C.Interaction of pectin with amino acids and other amino compounds in aqueous solution[J].Food hydrocolloids,2004,18(4):677-683.
[4]Fan L,Cao M,Gao S,et al.Preparation and characterization of a quaternary ammonium derivative of pectin[J].Carbohydrate Polymers,2012,88(2):707-712.
[5]Geresh S,Dawadi R P.Chemical modifications of biopolymers:quaternization of the extracellular polysaccharide of the red microalga Porphyridium sp[J].Carbohydrate Polymers,2000,43(1):75-80.
[6]Fan L,Gao S,Wang L,et al.Synthesis and anticoagulant activity of pectin sulfates[J].Journal of Applied Polymer Science,2012,124(3):2171-2178.
[7]Martinichen-Herrero J C,Carbonero E R,Gorin P A J,et al.Anticoagulant and antithrombotic activity of a sulfate obtained
from a glucan component of the lichen Parmotrema mantiqueirense Hale[J].Carbohydrate polymers,2005,60(1):7-13.
[8]Wang J,Zhang Q,Zhang Z,et al.Antioxidant activity of sulfated polysaccharide fractions extracted from Laminaria japonica[J].International Journal of Biological Macromolecules,2008,42(2):127-132.
[9]Rui hong Liang,Ling hua Wang,Jun Chen,et al.Alkylated pectin:Synthesis,characterization,viscosity and emulsifying properties[J].Food Hydrocolloids,2015,50:65-73.
[10]Gedye R,Smith F,Westaway K,et al.The use of microwave ovens for rapid organic synthesis[J].Tetrahedron letters,1986,27(3):279-282.
[11]Giguere R J,Bray T L,Duncan S M,et al.Application of commercial microwave ovens to organic synthesis[J].Tetrahedron letters,1986,27(41):4945-4948.
[12]de la Hoz A,Diaz-Ortiz A,Moreno A.Microwaves in organic synthesis.Thermal and non-thermal microwave effects[J].Chemical Society Reviews,2005,34(2):164-178.
[13]Fischer A,Houzelle M C,Hubert P,et al.Detection of intramolecular associations in hydrophobically modified pectin derivatives using fluorescent probes[J].Langmuir,1998,14(16):4482-4488.
[14]Pelletier S,Hubert P,Lapicque F,et al.Amphiphilic derivatives of sodium alginate and hyaluronate:synthesis and physico-chemical properties of aqueous dilute solutions[J].Carbohydrate Polymers,2000,43(4):343-349.
[15]Chen J,Wu S S,Liang R H,et al.The effect of high speed shearing on disaggregation and degradation of pectin from creeping fig seeds[J].Food chemistry,2014,165:1-8.
[16]Ma S,Wang Z.Pulsed electric field-assisted modification of pectin from sugar beet pulp[J].Carbohydrate polymers,2013,92(2):1700-1704.
[17]Pelletier S,Hubert P,Payan E,et al.Amphiphilic derivatives of sodium alginate and hyaluronate for cartilage repair:rheological properties[J].Journal of biomedical materials research,2001,54(1):102-108.
[18]Baississe S,Ghannem H,Fahloul D,et al.Comparison of structure and emulsifying activity of pectin extracted from apple pomace and apricot pulp[J].World Journal of Dairy & Food Sciences,2010,5(1):79-84.
[19]刘亚伟,邢伟亮,田景霞,等.微波干法制备高取代度阳离子淀粉的研究[J].粮食与饲料工业,2008(3):24-26.
[20]曹宝格,李华斌,罗平亚,等.疏水缔合水溶性聚合物AP4清水溶液的流变特征[J].油田化学,2006,22(2):168-172.
Microwave-assisted alkylation of pectin and its properties
FENG Si-jie,WANG Ling-hua,LIANG Rui-hong*,CHEN Jun,LIU Wei
(State Key Laboratory of Food Science and Technology,Nanchang University,Nanchang 330047,China)
In order to investigate the feasibility of microwave radiation for assisting alkylation of pectin,the influence of ratio of pectin to bromohexane,microwave power and radiation time on the degree of substitution(DS),as well as the physicochemical properties of alkylated pectin were studied.The results showed that the DS was increased first and reached a plateau value with the increase of the ratio,microwave power,and radiation time.When reaction took place at molar mass ratio of pectin to bromohexane 1∶1,and 320 W for 240 s,the DS reached 4.81%,which were superior to that obtained by 24 h traditional method(DS=3.60%).This results indicated that microwave radiation can improved the DS of alkylated pectin within a shorter processing time.With the increase of DS,microwave-assisted alkylated pectin had higher apparent viscosities,emulsifying capacity(EC)and emulsion stability(ES)compared with original pectin.The maximum EC and ES of microwave-assisted alkylated pectin could reach 57.29% and 93.61%,which was better than that of traditional prepared pectin and the original one.
alkylated pectin;synthesis;microwave radiation;apparent viscosities;emulsifying properties
2015-09-21
俸思洁(1990-),女,硕士研究生,研究方向:食品营养,E-mail:599730117@qq.com。
梁瑞红(1966-),女,博士,研究员,研究方向:天然产物的研发与开发,E-mail:liangruihong@ncu.edu.cn。
国家自然科学基金资助项目(31260386)。
TS201.1
A
1002-0306(2016)07-0052-05
10.13386/j.issn1002-0306.2016.07.002
猜你喜欢
食品工业科技(2021年23期)2021-12-16 02:21:14
当代化工研究(2021年22期)2021-04-11 18:12:22
中国化工贸易·上旬刊(2020年3期)2020-09-10 07:22:44
中国油脂(2020年7期)2020-07-14 11:13:48
精密成形工程(2020年3期)2020-06-08 12:04:42
中国粮油学报(2017年5期)2017-07-19 12:47:30
辽宁化工(2017年7期)2017-03-20 03:56:27
橡胶工业(2016年9期)2016-02-24 02:05:09
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
科技与创新(2015年20期)2015-10-29 23:36:33
