
浙江地区猪戊型肝炎病毒全基因组序列分析
2016-08-30 09:38帅江冰吴姗虞惠贞何永强张晓峰
质量安全与检验检测 2016年3期
帅江冰 吴姗 虞惠贞 何永强 张晓峰
(浙江省检验检疫科学技术研究院 浙江杭州 310016)
浙江地区猪戊型肝炎病毒全基因组序列分析
帅江冰吴姗虞惠贞何永强张晓峰*
(浙江省检验检疫科学技术研究院浙江杭州310016)
采用套式RT-PCR方法首次对浙江省猪群中戊型肝炎病毒的全基因组进行分段扩增,并对其两端非编码区采用末端快速扩增法进行扩增,T-A克隆测序后拼接分析。JH-14-03株全基因组共7253 bp,5’端至3’端依次为 5’非编码区(25 bp)、ORF1基因(5124 bp)、ORF3基因(339 bp)、ORF2基因(1983)及包含一个18个碱基A组成的Poly(A)尾的3’非编码区(86 bp)。JH-14-03株戊型肝炎病毒与已发表的基因4型戊型肝炎病毒全基因组核苷酸同源性最高,为85.6%-94.7%,显著高于与基因1、2、3型已发表戊型肝炎病毒全基因序列的同源性。分子进化分析显示JH-14-03株与大部分国内猪源和人源戊型肝炎病毒同属于戊型肝炎病毒基因4型毒株。
戊型肝炎病毒;全基因组序列分析
1 前言
戊型肝炎(Hepatitis E)由戊型肝炎病毒(Hepatitis E virus,HEV)引起,是一种经粪-口传播、以黄疸为主的急性传染病[1]。戊型肝炎的病死率较甲、乙、丙型和丁型肝炎高,其中患病孕妇的病死率可高达21%[2,3]。自1955年印度发生第1次大暴发以来,戊型肝炎已在亚洲、非洲与拉丁美洲的很多国家广泛流行[4],近年来亦在日本等发达国家散发流行[5]。我国是戊型肝炎高发区,新疆、辽宁、山东等省均有流行[3,6],对公共卫生和人类健康构成了极大威胁。
HEV基因组为单股正链RNA,约7.3 kb,依次包含5’非编码区、3个部分重叠的开放读码框(ORF)以及3’端非编码区和一个polyA尾[7]。自Meng等[8]1997年首次分离鉴定猪戊型肝炎病毒 (sHEV)以来,越来越多的研究表明猪源HEV与人源HEV基因组具有很高的同源性,在遗传进化关系上也有较高的亲缘性[9-10]。猪源HEV能通过人工接种感染黑猩猩等灵长类动物;人HEV也能成功感染猪等动物[11,12]。因此,戊型肝炎被认为是人畜共患性疾病[12,13]。种系进化研究表明不同地区来源的HEV基因组序列存在一定差异,可被划分成4个主要的基因型[14]。其中基因1型和2型仅分离于人类,大规模戊肝暴发流行均为此类HEV引起;基因3型和4型见于小规模流行和临床散发,能感染多种哺乳动物[15],并已有生食鹿肉和猪肉导致此类HEV感染的报道[16,17],此类HEV的主要天然宿主为猪。我国流行的HEV毒株则主要为1型和4型[18],近年来,在我国上海、浙江等地区的猪群中也陆续发现了HEV 3感染[19,20]。本研究在浙江省猪群中HEV流行分布及其特征分析的基础上,对来源于猪粪便中的JH-14-03 HEV毒株进行了全基因组扩增分析,以进一步分析浙江省HEV的基因结构、准确定型,便于深入了解解人源HEV与猪源HEV遗传进化关系。
2 材料与方法
2.1材料
2.1.1试剂
AmbionMagMAX-96TMViralRNAIsolationKit:Lifetechnologies,美国;SMART 5’RACE试剂盒:Clonetech,美国;DEPC水 (试剂纯)、Prime-ScriptTM 1st Strand cDNA Synthesis Kit、3'RACE试剂盒:大连宝生物公司。
2.1.2仪器
全自动核酸提取仪:Thermofisher Kingfisher Flex,美国;PCR仪:Biometra T3000,德国。
2.1.3样本来源
HEV病毒株JH-14-03:2014年采集自浙江省某规模化养殖场猪粪便,经RT-PCR检测为HEV RNA阳性。
2.2方法
2.2.1RNA提取
取1-2 g粪便样品,加入0.02 M PBS,彻底混匀制成20%的悬液(w/v);10000 r/min离心20 min,小心吸取上清,按Ambion MagMAX-96TM Viral RNA IsolationKit说明并使用全自动核酸提取仪提取RNA,最后加入40 μL DEPC水洗脱,-70℃保存备用。
2.2.2HEV全基因扩增
参照已发表的HEV各基因型序列,设计10对引物用于套式PCR分段扩增基因组序列(图1,表1)。提取的 RNA按 PrimeScriptTM 1st Strand cDNA Synthesis Kit说明书反转录为cDNA:首先取抽提的RNA 8 μL,分别加入表1中各扩增片段外侧引物的下游引物1 μL(20 μM)和dNTP 1 μL(10 mM),65℃处理5 min,立即冰浴。随后加入RT-buffer 4 μL,Rnasin 0.5 μL(40 U/μL),PrimeScript Rnase 1 μL (200 U/μL),双蒸水4.5 μL,42℃水浴1.5 h,70℃灭活15 min。直接取cDNA产物6 μL以各片段的外侧引物进行PCR扩增,条件为95℃ 5 min、94℃ 1min、52℃ 30 s、72℃ 45 s,35个循环,最后72℃ 10 min。取4 μL第一轮PCR产物以各片段的内侧引物进行第二轮PCR,反应体系和条件同第一轮PCR。
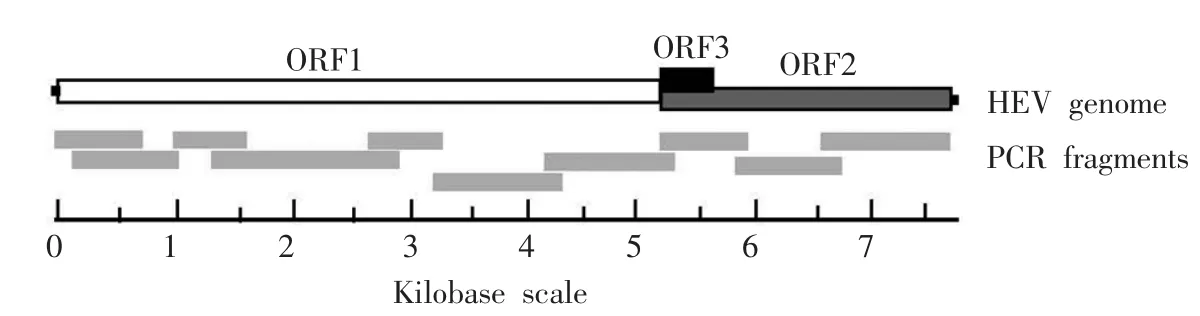
图1HEV全基因组分段PCR扩增示意图

表1 HEV全基因组套式PCR分段扩增引物及其碱基位点

(续表1)
2.2.3末端快速扩增法(RACE)
5’RACE按 CLONTECH 公 司 SMART5’RACE试剂盒说明进行,其中以片段1外侧引物作为基因特异性下游引物(表1)。
3’RACE按照TaKaRa试剂盒说明进行,用片段10引物作为基因特异性上游引物进行半套式反应。
2.2.4序列分析
分别将10段产物T-A克隆后送往上海英骏公司测序。测序结果拼接后,利用CLUSTAL X(1.83)将其与国内外其他地区的HEV毒株ORF2序列进行多重比对分析,然后分析不同基因片段中发生突变的位点。利用软件 MolecularEvolutionary Genetics Analysis(MEGA,version 3.1)进行分子进化关系分析。
HEV氨基酸位点的选择压力通过 MEGA (version 5.1)软件在Muse-Gaut密码子替换模型的基础上,以最大似然法进行进化分析。以标准化的非同义突变(dN)和同义突变(dS)的差值作为进化压力的判断指标,即当dN-dS大于0表示位点发生的是正性选择;若dN-dS小于0表示位点发生的是负性选择;dN-dS等于0表示位点受中性选择的作用。同时对位点突变是否符合中性突变进行统计学检验,若p大于0.05则符合零假设,即位点的突变符合中性突变。
3 结果
3.1HEV基因结构
猪源HEV 4毒株JH-14-03株全基因组共7253 bp,5’端至3’端分别为:25个核苷酸组成的5’非编码区(1-25 nt),全长5124 bp的ORF1基因(26-5149 nt),339 bp的ORF3基因(5174-5512 nt),1983 bp的ORF2基因(5185-7167 nt)以及一段86 bp的包括18个碱基A组成的PolyA尾的3’非编码区 (7167-7253 nt)。JH-14-03株HEV ORF1和ORF2基因不重叠,ORF3与ORF2基因部分重叠。
3.2HEV基因序列分析
将JH-14-03全基因组与Genebank中已发表的HEV全序列比对,结果显示JH-14-03株HEV与已发表的基因4型HEV全基因组苷酸同源性较高,为85.6%-94.7%(表2),显著高于与基因1、2、3型已发表HEV全基因序列的同源性。从ORF序列比对结果来看,ORF1相对最为保守,大部分毒株间ORF1同源性要高于ORF2和ORF3基因同源性。

表2 HEV JH-14-03分离株与已发表的不同型HEV全基因组序列比对分析(%)
JH-14-03毒株ORF1基因编码1708个氨基酸,与4型HEV ORF1同源性较高,部分毒株可高达90%以上(表2)。进一步结合已发表的HEV全基因序列对ORF1核苷酸点突变进行分析,结果表明,ORF1点突变位点较为均衡的散步分于整个基因中,其中以3000-3500 nt区域发生频率最高。选择压力分析显示HEV ORF1虽然有较多位点dN-dS值大于0(图2),但均接受零假设(p大于0.05),其进化压力以中性选择为主。
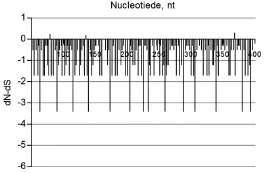
图2 HEV 4 ORF 1基因氨基酸位点dN-dS值(1-400 aa)
JH-14-03株ORF 2基因序列均为1983 bp,与HEV 1、HEV 2、HEV3和HEV4参考序列的核苷酸同源性分别73.5-75.3%、71.2%、73.4-76.3%、5.7-91.6%,与HEV 4型参考序列同源性最高。JH-14-03株ORF 2编码661个氨基酸,与已发表的HEV 4毒株进行选择压力分析其后发现其中有2个位点dN-dS值大于0(图3):第67位的赖氨酸,dN-dS为0.29;第208位的苯丙氨酸,dN-dS为0.16,但统计学分析表明p分别为0.356和0.529,因此,不能认为这两个位点在进化时受正性选择的作用。其余大多数氨基酸位点的dN-dS值小于0,但其统计学检验结果为零假设 (p大于0.05)。综上所述,HEV 4 ORF2蛋白位点同义替换率和非同义替换率相似,其进化压力以中性选择为主。

图3 HEV 4 ORF2基因氨基酸位点dN-dS值(1-228 aa)
JH-14-03毒株ORF 3基因含339个核苷酸,编码112个氨基酸,与部分日本人源HEV和中国猪源HEV一致,但与大多数人源HEV和部分中国猪源HEV相比少了一个编码氨基酸。
3.3非编码区序列分析
已报道的HEV全基因组5’编码区序列从9个碱基到33个碱基不等,但未见特征性报道。JH-14-03的5’非编码区分别为25 bp,比对后显示各地区均有与本研究毒株相似类型的HEV毒株。JH-14-03的3’非编码区不包括PolyA尾为68 bp,与已发表HEV毒株3’非编码区同源性比对结果与全基因组或ORF同源性比对结果一致,即JH-14-03与4 型HEV同源性高。JH-14-03的3’非编码区还含有18个PolyA尾,有报道称RNA病毒的PolyA尾长短与病毒致病性强弱可能存在一定的关系,但具体相关性还未见清晰证明。
3.4全基因组分型
将JH-14-03全基因组序列与其他各型HEV毒株序列比对后构建进化树,结果显示可以分为明显的4支。JH-14-03分布于4型分支中,与中国猪源HEV毒株亲缘性较近。与以ORF2构建的进化树一致,日本毒株构成了相对独立的一个小分支,表现出一定的地区聚集性。进化树分型显示猪源HEV与人源HEV均散布于3型和4型分支中,亲缘性较高,未出现明显的种间聚集。
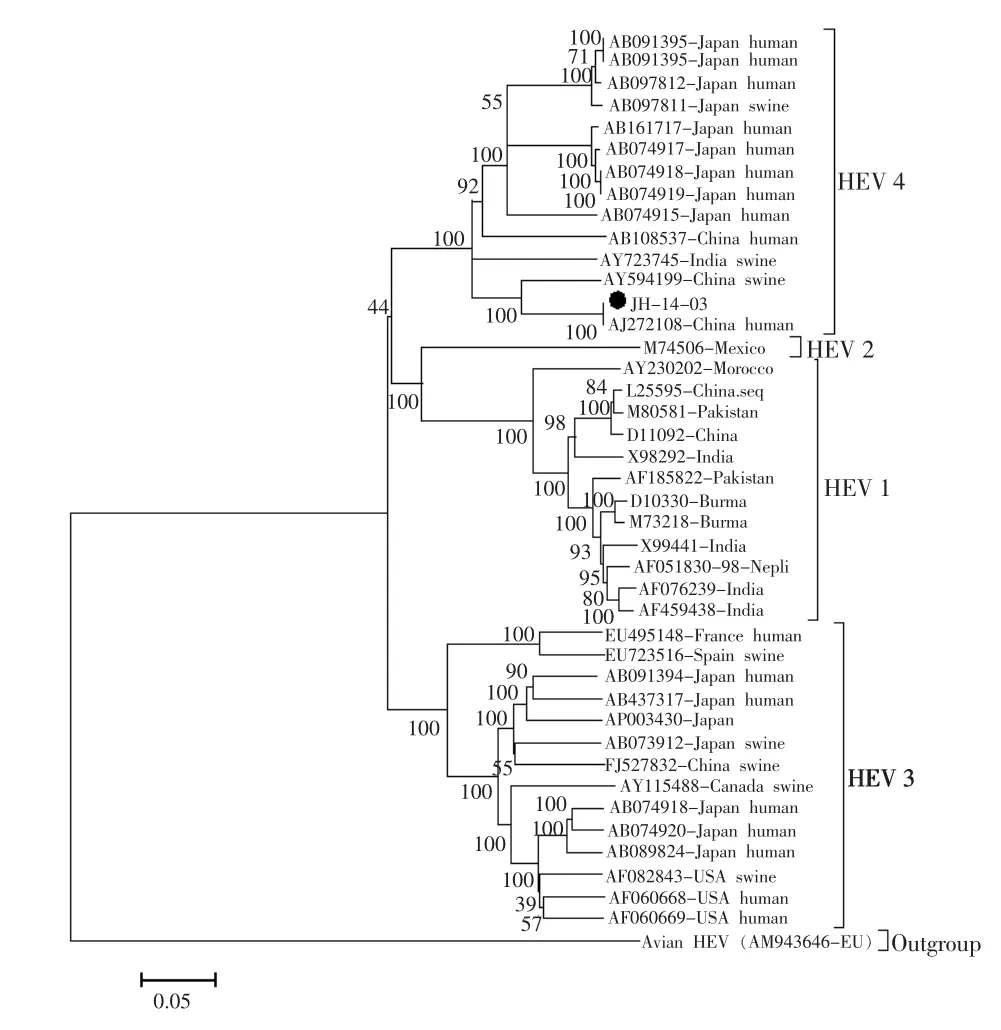
图4 HEV JH-14-03分离株全基因序列分子进化关系分析
4 讨论
本研究首次报道了浙江省猪源HEV JH-14-03株全基因组序列。与已报道的其他猪源或人源HEV分离株全序列进行比较后发现,JH-14-03株与HEV 4同源性最高,但也存在大量的核苷酸突变。JH-14-03全基因组包含两个非编码区和3个主要ORF,其基因结构均与大部分HEV 4一致。对ORF1 和ORF 2基因每个氨基酸位点的选择压力分析发现,虽然有部分位点的dN-dS值大于0,但均无统计学意义,因此本研究认为HEV 4 ORF1和ORF2在进化过程中主要受中性选择的作用。由于同义替换不受自然选择作用,所以在排除其他干扰因素的情况下,许多基因位点的同义替换速率往往相似且保持稳定。
病毒基因组不同位点按照不同的突变结果,表现出不同的突变率。HEV基因的突变以第三密码子的同义突变为主,这可能由于非同义突变的位点往往受到功能限制而导致其突变率较低。但由于病毒的迁移、急性散发和重组适应等进化因素,造成了更多位点的同义突变。同时不同基因型由于宿主范围的原因,其突变率也有所差别。由于动物宿主的生长周期要明显短于人类,因此存在于动物宿主的HEV的生存压力要大于人,导致其快速突变,以增强适应性,进而导致HEV 3和HEV 4的突变率要高于HEV 1。
本研究对HEV全基因组分型分析后显示JH-14-03株分布于HEV 4型分支中,与核苷酸同源性分析结果一致。分子进化分析显示只有部分日本HEV毒株显示了一定的地区聚集性,其余不同地区的人源和猪源HEV均散布不同型分支中,未表现出明显的地区差异,提示大部分地区HEV毒株尚未进化形成明显的特征,但也可能是HEV毒株跨地区迁移、传播的结果。猪源HEV与人源HEV均散布于3型和4型分支中,亲缘性较高,未出现明显的种间聚集,提示HEV在人、猪等物种间跨物种传播的可能性[12,13]。有报道还显示有人因食用了野猪肉和鹿肉而引起急性戊肝[16,17],说明戊型肝炎病毒可能在自然状态下或通过食物等在动物和人之间传播。此外,与猪频繁接触的人群(如饲养员、兽医、屠工)中戊型肝炎病毒的抗体水平明显高于正常人群,也提示猪HEV可能交叉感染人类[11]。因此,HEV被认为在某些地区可跨越种间屏障,相互传播,但传播机制如何,还有待于进一步研究。
5 结论
本研究首次分析了浙江省猪群中基因4型戊型肝炎病毒的全基因组序列,全长7253 bp,包含两个非编码区和3个主要ORF,其基因结构与大部分HEV 4一致,但存在大量的核苷酸突变。
[1] Arankalle L P,Chobe J Jha,Chadha M S,et al.Aetiology of acute sporadic non-A,non-B hepatitis in western India[J]. Med Virol,1993,40:121-125.
[2] Tam W,Smith M M,GUERRA M E,et al,Hepatitis E virus (HEV):molecular cloning and sequencing of the full-length viral genome[J].Virology,1991,185:120-131.
[3]王凤阳,杜丽,张莉娜.猪戊型肝炎病毒的研究进展[J].中国热带医学,2007,7(6):1000-1004.
[4]Gardinali N R,Barry A F,da Silva P F,et al.Molecular detection and characterization of hepatitis E virus in naturally infected pigs from Brazilian herds[J].Res Vet Sci,2012,93:1515-1519.
[5]Wenzel J J,Preiss J,Schemmerer M,et al.Detection of hepatitis E virus(HEV)from porcine livers in Southeastern Germany and high sequence homology to human HEV isolates[J].J Clin Virol,2011,52:50-54.
[6]庄辉,毕胜利,王佑春,等.我国戊型肝炎研究[J].北京大学学报(医学版),2002,5(34):434-439.
[7] Purdy M A.olution?of the?hepatitis E virus?polyproline?region:?order?from?disorder[J].J Virol,2012,86(18):10186-93.
[8]Meng X J,Purcell R H,Halbur P G,et al.A novel virus in swine is closely related to the human hepatitis E virus[J]. Pros Natl Acad Sci USA,1997,94:9860-9865.
[9]Wang Y C,Zhang H Y,Xia,et al.Prevalence,isolation,and partial sequence analysis of hepatitis E virus from domestic animals in China[J].J Med Virol,2002,67:516-521.
[10]Nakamura M,Takahashi K,Taira K,et al.Hepatitis E virus infection in wild mongooses of Okinawa,Japan:demonstration of anti-HEV antibodies and a full-genome nucleotide sequence [J].Heaptol Res,2006,34:137-140.
[11]Balayan M S,Usmanov R K,Zamyatina N A,et al.Brief report:experimental hepatitis E infection in domestic pigs[J].J Med Virol,1990,32:58-59.
[12]Song Y J,Park W J,Park B J,et al.Hepatitis E virus infections in humans and animals[J].Clin Exp Vaccine Res,2014,3 (1):29-36.
[13]Meng X J.Hepatitis E virus:animal reservoirs and zoonotic risk [J].Vet Microbiol,2010,140:256-265.
[14]Li T C,Yang T,Shiota T,et al.Mlecular detection of hepatitis E virus in rivers in the Philippines[J].Am J Trop Med Hyg,2014,90(4):764-6.
[15]OKAMOTO H.Genetic variability and evolution of hepatitis E virus[J].Virus Research,2007,127:216-228.
[16]Tei S,Kitajima N,Takahashi K,et al.Zoonotic transmissionof hepatitis E virus from deer to human beings[J].Lancet.2003,362:371-373.
[17]Masuda J I,Yano K,Tamada Y,et al.Acute hepatitis E of a man who consumed wild boar meat prior to the onset of illness in Nagasaki.Japan[J].Hepatol Res,2005,31(3):178-183.
[18]郭清顺,葛胜祥,熊君辉,等.戊型肝炎病毒基因1型和基因4型中和表位区域分子差异研究[J].病毒学报,2007,23(6):454-458.
[19]Si F S,Zhu Y M,Dong S J,et al.Full genomic sequence analysis of swine genotype 3 hepatitis E virus isolated from Shanghai[J].Virus Res,2009,144(1-2):290-293.
[20]张晓峰,帅江冰,李爱云,等.猪戊型肝炎病毒荧光定量PCR检测方法的建立及其分子流行病学 [J].中国兽医学报,2011,31 (6):822-827.
Full Genome Sequence Analysis of Swine Genotype 4 Hepatitis E Virus Isolated from Zhejiang
SHUAI Jiangbing,WU Shan,YU Huizhen,HE Yongqiang,ZHANG Xiaofeng*
(Zhejiang Academy of Science and Technology for Inspection and Quarantine,Hangzhou,Zhejiang,310016)
The full-length genomic sequence of a swine hepatitis E virus(HEV)isolate(JH-14-03)isolated from feces of pig in Zhejiang province was determined.The complete genome of 7253 nucleotides consisted of 5’non-coding region,ORF1,ORF3,ORF2 and 3’non-coding region that including poly(A)tail of 18 residues.Comparative full-length genome sequence revealed that sequence of JH-14-03 was closely related to other HEV isolates published in NCBI with identity of 85.6%-94.7%.Phylogenetic analysis suggested that the isolate JH-14-03 clustered in group 4 with most of published swine and human HEV isolates.
Hepatitis E Virus;Full Genome Sequence Analysis
S851.34
E-mail:sjb@ziq.gov.cn;*通信作者Email:zxf@ziq.gov.cn
浙江省重点研发计划项目(2015C02G4130003);国家质检总局科技计划项目 (2013IK046)
2015-12-28
猜你喜欢
今日农业(2022年14期)2022-09-15
青春期健康(2022年14期)2022-08-02
南方农业学报(2022年4期)2022-07-14
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
青春期健康(2021年14期)2021-08-03
西部医学(2021年7期)2021-07-26
猪业科学(2021年3期)2021-05-21
中国生殖健康(2019年7期)2019-01-06
中国猪业(2017年11期)2017-12-11
