
一种咔唑衍生物结构及红外光谱的理论研究
2016-08-11 08:15:39吴世永王玉良
新技术新工艺 2016年7期
李 慧,吴世永,王玉良
(海军航空工程学院 基础部,山东 烟台 264001)
一种咔唑衍生物结构及红外光谱的理论研究
李慧,吴世永,王玉良
(海军航空工程学院 基础部,山东 烟台 264001)
应用Gaussian03W软件,通过密度泛函理论B3LYP/6-31G(d)方法对9-甲基丙烯酰氨咔唑分子的结构进行优化,得到了其最稳定构型并计算出了该化合物分子的键长、键角和空间二面角等数据。在优化的最稳定构型的基础上,以B3LYP/6-31G为基组,用同样的计算方法得到了该分子的红外振动频率,并绘制出了红外光谱图,通过频率分析说明优化得到的9-甲基丙烯酰氨咔唑分子的结构稳定合理。最后,对9-甲基丙烯酰氨咔唑分子的红外光谱峰位进行了归属分析。
9-甲基丙烯酰氨咔唑;红外光谱;Gaussian03;密度泛函理论
咔唑及其衍生物具有刚性稠环结构,强的分子内电子转移、优良的空穴传输性使得这类含氮芳杂环化合物表现出许多丰富的功能特性,从小分子到高分子使得咔唑及其衍生物在光电材料、医药和农药等多领域具有潜在的广泛应用[1-5]。近年来,对于咔唑及其衍生物结构与性质的研究十分活跃,引起了人们的广泛关注。
红外光谱法是对各种吸收红外光的化合物进行定性和定量分析的一种方法,是研究分子振动光谱和分子结构的重要手段[6-11]。而Gaussian03W是一种常用的量子化学计算软件,它可以搭建化学物质空间几何优化结构,给出能量值,并计算出相应的红外光谱、拉曼光谱以及核磁共振等数据。本文运用Gaussian03W计算程序,应用密度泛函理论(DFT),以B3LYP/6-31G(d)为基组,得到了9-甲基丙烯酰氨咔唑的最稳定构型,并采用同样方法,以B3LYP/6-31G为基组,计算了该分子的振动频率,绘制了其红外光谱图。
1 计算方法
采用GaussianView程序构建了9-甲基丙烯酰氨咔唑的初始构型。在Gaussian03W程序包中采用量子化学密度泛函理论方法,以B3LYP/6-31G(d)为基组,对这种化合物的空间构型进行全结构优化,得到了该化合物的最稳定态构型构,分子的键长、键角和空间二面角等数据。在优化的稳定构型的基础上,用同样的方法,以B3LYP/6-31G为基组,计算了该分子的红外振动频率,绘制了红外光谱图,并对其红外光谱峰位进行了合理的归属。
2 结果与讨论
2.1分子的稳定态构型
通过密度泛函理论B3LYP/6-31G(d)方法,计算优化9-甲基丙烯酰氨咔唑分子的结构,使得分子能量达到最小值,几何结构最稳定,才能进行下一步计算振动频率。优化后的立体结构、原子编号以及Cartesian坐标如图1所示。优化后稳定态分子的键长、键角及二面角数据见表1。

图1 9-甲基丙烯酰氨咔唑分子优化后的稳定构型
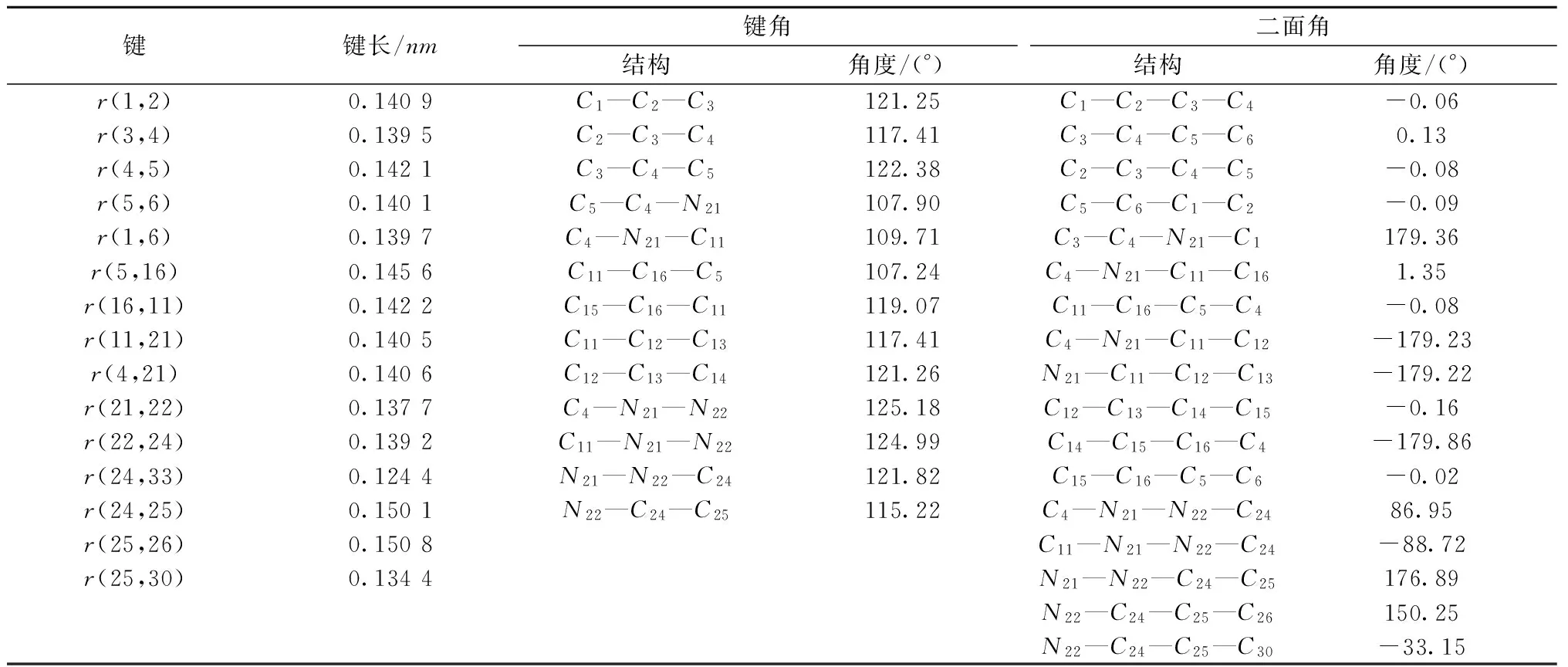
键键长/nm键角结构角度/(°)二面角结构角度/(°)r(1,2)0.1409C1—C2—C3121.25C1—C2—C3—C4-0.06r(3,4)0.1395C2—C3—C4117.41C3—C4—C5—C60.13r(4,5)0.1421C3—C4—C5122.38C2—C3—C4—C5-0.08r(5,6)0.1401C5—C4—N21107.90C5—C6—C1—C2-0.09r(1,6)0.1397C4—N21—C11109.71C3—C4—N21—C1179.36r(5,16)0.1456C11—C16—C5107.24C4—N21—C11—C161.35r(16,11)0.1422C15—C16—C11119.07C11—C16—C5—C4-0.08r(11,21)0.1405C11—C12—C13117.41C4—N21—C11—C12-179.23r(4,21)0.1406C12—C13—C14121.26N21—C11—C12—C13-179.22r(21,22)0.1377C4—N21—N22125.18C12—C13—C14—C15-0.16r(22,24)0.1392C11—N21—N22124.99C14—C15—C16—C4-179.86r(24,33)0.1244N21—N22—C24121.82C15—C16—C5—C6-0.02r(24,25)0.1501N22—C24—C25115.22C4—N21—N22—C2486.95r(25,26)0.1508C11—N21—N22—C24-88.72r(25,30)0.1344N21—N22—C24—C25176.89N22—C24—C25—C26150.25N22—C24—C25—C30-33.15
由图1可以看出,9-甲基丙烯酰氨咔唑分子具有C1对称性,其中咔唑基团与甲基丙烯酰氨基不在同一平面上,二者之间有一定的二面角。图1和表1的计算结果显示,咔唑部分在同一平面上,C3—C4—C5所在的苯环、C16—C11—C12所在的苯环以及C4—N21—C11所在的五元环其二面角的绝对值都近似等于0°或180°。而二面角C4—N21—N22—C24和C11—N21—N22—C24分别为86.95°和-88.72°,这说明甲基丙烯酰氨与咔唑不在同一平面上。键长数据显示,r(1,2)、r(4,5)分别为0.140 9和0.142 1nm,均比单个苯环的键长要长;r(3,4)、r(1,6)分别为0.139 5和0.139 7nm,均比单个苯环的键长短;r(5,16)= 0.145 6nm,比正常碳碳单键短;r(4,21)、r(11,21)分别为0.140 6和0.140 5nm,均比正常碳氮单键短。这些数据说明咔唑部分形成了较大的共轭体系。
2.2理论计算的振动频率及红外光谱
用Gaussian03W软件计算得到的9-甲基丙烯酰氨咔唑的红外振动频率见表2。对频率数据进行分析得到3个最小振动频率分别为33.07(即1.1×105Hz)、40.50和44.43 /cm,3个最小吸收强度分别为0.558、1.348和2.131km/mol。由此可以看出,其振动光谱中未出现虚频,说明优化得到的构型合理。
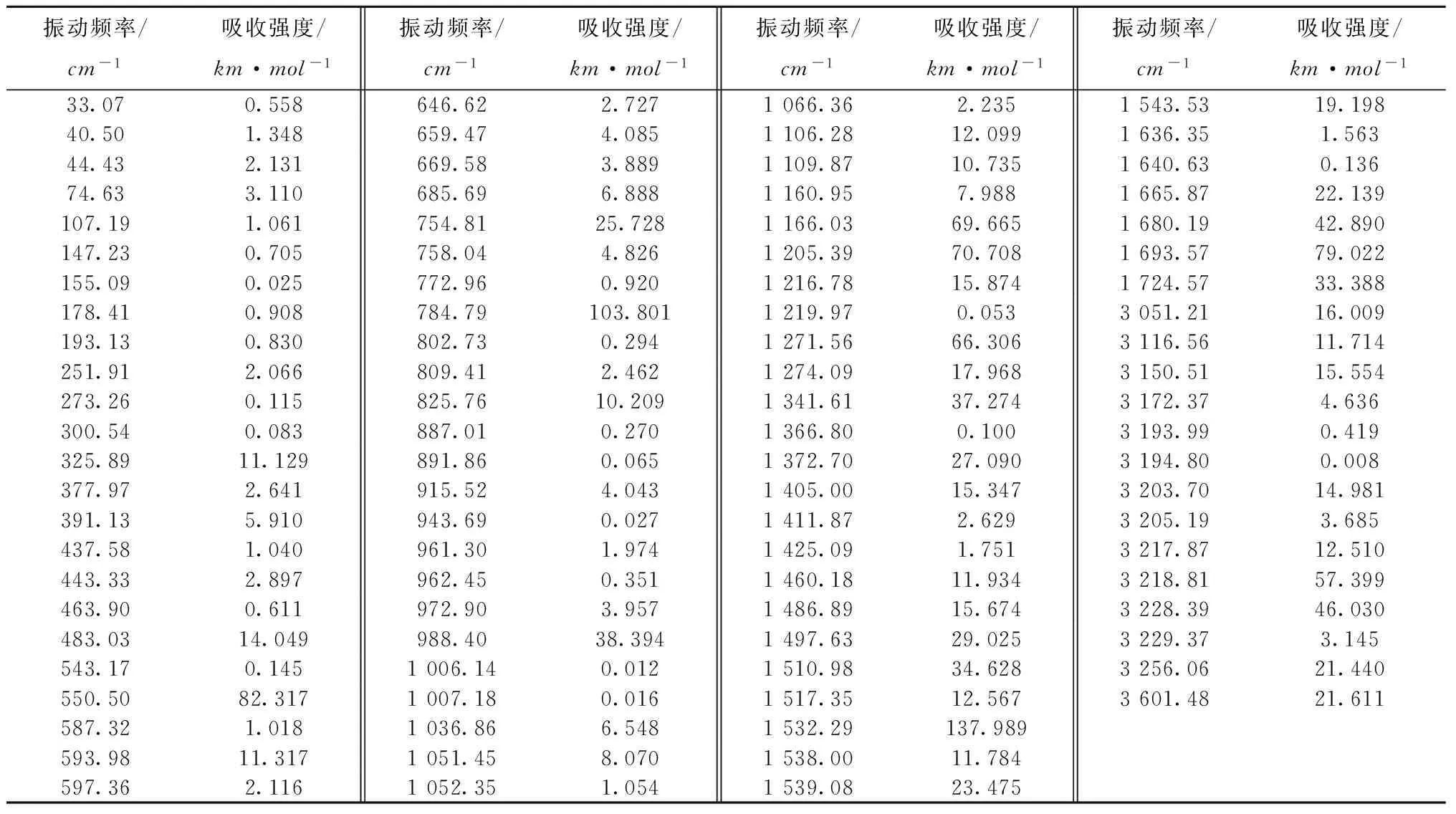
表2 9-甲基丙烯酰氨咔唑的红外振动频率
9-甲基丙烯酰氨咔唑的红外光谱计算结果如图2所示。红外光谱对应分子的结构和化学键,本文应用GaussianView程序对9-甲基丙烯酰氨咔唑的红外光谱进行了归属。振动频率在3 601 /cm处的较弱吸收峰对应的是N—H的伸缩振动;3 256 /cm处的较弱吸收峰对应的是—C=H2上的碳氢不对称伸缩振动;在3 203~3 228 /cm内的较弱吸收峰对应的是苯环上的碳氢不对称伸缩振动特征峰;在1 693、1 724 /cm处的吸收峰对应的是甲基丙烯酰氨片段上的碳碳对称弯曲振动;在1 665、1 680 /cm处的较强吸收峰对应的是咔唑片段苯环上的C—H面内弯曲振动;在1 532 /cm处的较强吸收峰对应的是N—H弯曲振动;在784 /cm处的较强吸收峰是苯环上的C—H面外弯曲振动。分子的红外光谱起源于分子的振动基态与振动激发态之间的跃迁,只有在跃迁的过程中有偶极矩变化的才会出现红外光谱[12-13]。上述数据表明,标题分子振动过程中偶极矩变化较大,红外振动活性强。

图2 9-甲基丙烯酰氨咔唑的红外光谱计算结果
3 结语
采用GaussianView程序构建了9-甲基丙烯酰氨咔唑的初始构型。在Gaussian03W程序包中采用量子化学密度泛函理论方法,以B3LYP/6-31G(d)为基组,对这种化合物的空间构型进行了全结构优化,得到了该化合物分子的键长、键角和空间二面角等数据,表明在一般条件下此化合物还是很稳定的。在优化的稳定结构的基础上,用同样的方法,以B3LYP/6-31G为基组,计算了该分子的红外振动频率,绘制了红外光谱图,在频率值的计算结果中没有出现虚频,说明优化得到的9-甲基丙烯酰氨咔唑分子的构型合理,并对其红外光谱峰位进行了归属指认。目前,在Gaussian03W程序包中采用量子化学密度泛函理论的方法,所得到的计算结果是比较精确的,计算结果可以较好地应用于实践当中。本文的研究对9-甲基丙烯酰氨咔唑及其衍生物分子的红外光谱的实际测量和分析是一个可靠的技术参考。
[1] 苏玉苗,林海娟,李文木.咔唑及其衍生物在蓝光OLED中的应用[J].化学进展,2015,27(10):1384-1399.
[2]HeQY,LaiWY,MaZ,etal.Novelbluelight-emittinghyperbranchedpolyfluorenesincorporatingcarbazolelinkedstructure[J].Eur.Polym.J., 2008, 44:3169-3176.
[3] 张飞飞,周成合,颜建平.咔唑类化合物研究新进展[J].有机化学,2010,30(6):783-796.
[4] 梁利岩,张静娴,梅群波,等.含咔唑基聚合物的合成、发光性能及应用[J].精细化工,2007,24(8):743-746.
[5] 耿凤华,王永祥,刘保霞.咔唑及其衍生物在光电材料中的应用研究[J].应用化学,2011,40(8):1444-1450.
[6] 贾廷见,李朋伟,尚治国,等.糠醛分子的拉曼光谱和红外光谱研究[J]. 光散射学报, 2007,19(1):1-5.
[7] 郑纯智,张国华.溴化苄的合成工艺研究[J].广州化学,2005,30(3):22-27.
[8] 宋志国,王敏,姜恒,等.溴化钾-浓硫酸法合成苄基溴[J].工业催化,2008,16(1):48-53.
[9] 肖翠玲,徐文国,冷启贞.SF5CF3几何构型及红外振动光谱的量子化学研究[J]. 北京理工大学学报, 2005,25(5):462-469.
[10] 李晓明,张来斌,周留柱,等.2-吡啶甲醇红外光谱的密度泛函理论研究[J]. 光谱学与光谱分析, 2012,32(9):2358-2363.
[11] 孔轶众,孔祥和,李晓明,等. 二氯苯 3种同分异构体及其阳离子的红外光谱研究[J].光谱实验室,2009,26(6):1437-1441.
[12] 王庆学, 魏彦锋, 朱建妹, 等.B+注入HgCdTe外延材料的红外透射光谱分析[J]. 光子学报, 2005, 34(8):1179-1182.
[13] 林继鹏; 刘君华. 基于吸收峰混叠的红外混合气体分析方法的研究[J].ActaPhotonicaSinica, 2006, 35(3):408-412.
责任编辑郑练
TheoreticalResearchonStructureandInfraredSpectraoftheCarbazoleDerivative
LIHui,WUShiyong,WANGYuliang
(DepartmentofBasicSciences,NavalAeronauticalandAstronauticalUniversity,Yantai264001,China)
TheoptimizationoftheN-carbazol-9-ylmethacrylamideisperformedbydensityfunctionaltheorymethodatB3LYP/6-31G(d)oftheorywithGaussian03Wsoftwaretoobtainthemoststableconfigurationandcalculatethemolecularbondlength,bondangleandspacedihedralangle.Basedontheoptimizedconfiguration,theinfraredvibrationalfrequenciesandinfraredspectraareobtainedbythesamemethodatB3LYP/6-31G,throughthefrequencyanalysis,optimizedconfigurationoftheN-carbazol-9-ylmethacrylamideisstableandreasonable.Finallythepeakpositionoftheinfraredspectraisanalyzed.
N-carbazol-9-ylmethacrylamide,infraredspectra,Gaussian03,densityfunctionaltheory

O657.2A
李慧(1981-),女,讲师,主要从事量子化学及功能材料等方面的研究。
2016-02-16
猜你喜欢
扬子江诗刊(2023年3期)2023-05-06 10:40:14
大众文艺(2022年16期)2022-09-07 03:08:04
辽宁化工(2022年8期)2022-08-27 06:03:04
中学生数理化(高中版.高考数学)(2022年2期)2022-04-26 14:05:00
石油炼制与化工(2021年12期)2021-12-14 06:26:38
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:40
中学生数理化(高中版.高考数学)(2020年3期)2020-05-25 06:53:20
农药科学与管理(2019年5期)2019-08-13 00:48:02
高中生·天天向上(2018年1期)2018-04-14 09:24:38
塑料助剂(2018年6期)2018-03-25 05:59:16
