
功能性纳米羟基磷灰石的制备及其表征
2016-08-10 08:18赵颜忠张海斌周科朝
中国有色金属学报 2016年6期
赵颜忠,杨 敏,张海斌,朱 军,周科朝
(1. 中南大学 湘雅三医院,长沙 410013;2. 中南大学 粉末冶金国家重点实验室,长沙 410083;3. 中南大学 医用材料与器械研究中心,长沙 410013)
功能性纳米羟基磷灰石的制备及其表征
赵颜忠1, 2, 3,杨 敏1,张海斌2,朱 军2,周科朝2
(1. 中南大学 湘雅三医院,长沙 410013;2. 中南大学 粉末冶金国家重点实验室,长沙 410083;3. 中南大学 医用材料与器械研究中心,长沙 410013)
以硝酸钙、磷酸氢二氨等为反应原料,采用水热合成法制备羟基磷灰石(HAP)纳米颗粒,对该HAP颗粒以及添加精氨酸(Arg)或掺杂少量稀土离子铽(Tb)/铕(Eu)对 HAP颗粒的形貌修饰等进行研究,采用透视电镜(TEM)、X射线衍射仪(XRD)、傅立叶红外光谱仪(FTIR)等,对制备样品的结晶性、粒度、分散性等进行分析测试。结果表明:精氨酸表面修饰改变HAP纳米颗粒的表面Zeta电位,从而在一定程度上抑制HAP的生长速率;少量稀土离子Eu/Tb掺杂并不影响HAP/Arg纳米颗粒产物的结构,均为单一的HAP物相,所合成产物的粒径也为纳米级。经稀土金属铽/铕掺杂的精氨酸表面修饰的HAP纳米颗粒可作为基因转染载体。
羟基磷灰石;水热合成;精氨酸修饰;稀土掺杂
纳米级羟基磷灰石(Hydroxyapatite, HAP)首先由美国牙科医师BROWN和CHOW[1]于1987年首先发现,是最常见的一种生物活性材料,与人体硬组织的无机组分相似,具有良好的化学稳定性和生物相容性以及生物活性,对人体无毒副作用,在生物医学领域应用广泛[2]。随着纳米技术的兴起,人们对羟基磷灰石的研究热点逐渐向纳米领域发展,研究发现,HAP的许多特性与其粒径大小密切相关[3]。与普通的HAP相比,HAP纳米颗粒表现出独特的生物特性,如比表面积和表面能较大、溶解度较高、以及能够结合抗癌药物、核酸和蛋白质等更强的生物活性[4-5]。此外,HAP纳米颗粒具有抗癌活性,能抑制癌细胞的生长[6]。因此,对HAP纳米颗粒的制备及其临床医学应用的研究已越来越受到广大科研工作者的密切关注。同时,国内外研究发现[7-10],HAP纳米颗粒作为基因载体,还在于制备工艺简单,并易改性修饰、可以负载不同基因、浓缩保护基因并缓慢释放、生物稳定性好等优点。研究表明,HAP纳米颗粒是一种理想的基因载体材料。但是,这种纳米载体材料的基因转染率与其尺寸形貌和表面状态的关系以及基因转染机制仍有待深入研究[11]。因此,进一步探讨HAP纳米颗粒的转染机制及改进制备工艺以提高其转染效率是促使HAP纳米颗粒作为基因载体应用于临床基因治疗的关键。
为了阐释 HAP纳米颗粒的细胞跨膜转运途径及与细胞相互作用机制,实时动态观察HAP/Arg纳米颗粒在细胞内的跨膜转运入胞过程。本文作者采用硝酸钙、磷酸氢二氨等为反应原料,对水热合成法制备的HAP纳米颗粒以及添加精氨酸(Arginine)对HAP颗粒的形貌修饰等进行了研究,对制备样品的结晶性、粒度、分散性等进行分析测试。同时,探讨通过水热法合成一种新型改性的非病毒载体、稀土金属铽/铕掺杂的精氨酸修饰的HAP纳米颗粒,并进行表征,确认其适用于基因转染载体领域的应用。
1 实验
1.1 实验材料
本实验中采用的实验材料主要如下:硝酸钙Ca(NO3)2·4H2O(AR,中国医药集团上海化学试剂公司生产),磷酸铵(NH4)3PO4·3H2O(AR,中国医药集团上海化学试剂公司生产),磷酸氢钠Na2HPO4(AR,中国医药集团上海化学试剂公司生产),精氨酸(Arginine,Sigma公司生产),氧化铕Eu2O3(99.995%,湖南稀土金属研究院生产),氧化铽Tb2O3(99.995%,湖南稀土金属研究院生产),浓氨水(AR,山东滕州祥润化工有限公司生产),浓硝酸(AR,南京章扬石化有限公司生产),以及自行配制的磷酸缓冲盐溶液(PBS)和HEPES缓冲盐溶液(HBS)。
1.2 HAP纳米颗粒制备
取5.900 g的Ca(NO3)2·4H2O置于烧杯中,向烧杯中加蒸馏水至30 mL,充分搅拌形成澄清透明的溶液,再添加蒸馏水稀释至50 mL。称取1.447 g质量分数不小于85%的磷酸溶液,用蒸馏水稀释至30 mL。测得其pH值在1左右,用氨水调节其pH值至10,最后将溶液稀释至 50 mL。将磷酸铵溶液瞬间加入Ca(NO3)2溶液中,充分搅拌,溶液呈白色乳浊状,并用浓氨水调节溶液的pH值至9~10。将溶液倒入三口烧瓶中,固定在支架台上。烧瓶下部分浸入80 ℃的水液中,使外部水面高过烧瓶内部反应液面。调节搅拌器,以适当的速率使反应液充分混合。刚开始搅拌时液体为乳白色,此状态维持到整个陈化过程结束。将含混合液搅拌混匀后转入到水热釜中,然后将密封的高压釜置于恒温干燥箱中,在160 ℃下持续反应3 h。取出高压釜冷却后,取其中的沉淀,用去离子水和无水乙醇洗涤数次,然后真空干燥,即得到HAP纳米颗粒样品。
1.3 精氨酸修饰HAP纳米颗粒制备
取8.710 g的精氨酸与5.900 g 的Ca(NO3)2·4H2O置于烧杯中,加蒸馏水至30 mL,充分搅拌,可使液体分层。下层为澄清透明的溶液,上层为白色悬浮液。再将磷酸铵溶液瞬间加入含Ca2+精氨酸溶液,充分搅拌,调节溶液的 pH值至 9。将混合液倒入三口烧瓶中,浸入 80 ℃的水液中充分搅拌混合,此过程进行16 h。刚开始搅拌时,反应液面有少量白色泡沫产生,液体为乳白色。此状态维持到整个陈化过程结束。陈化过后将混合液倒入高压反应釜中,置于160 ℃的干燥箱中反应3 h,然后取出自然冷却。用去离子水和无水乙醇对反应的沉淀产物洗涤数次,经过真空干燥并研磨得到精氨酸修饰的HAP/Arg纳米颗粒样品。
1.4 HAP/Arg纳米颗粒的稀土掺杂
取2.0711 g氧化铕(Eu2O3)溶于15 mL去离子水中,用浓硝酸配制体积比1:1的硝酸盐溶液中,加热并搅拌使Eu2O3完全溶解,然后加入一定量的去离子水重复加热过程直到得到较为纯净的稀土硝酸铕(Eu(NO3)3)溶液,然后待Eu(NO3)3溶液冷却至室温后转入 200 mL的容量瓶中并定容配置成体积分数为0.0588mol/L的Eu(NO3)3溶液,最后将其储存在广口瓶中放于阴凉处保存,以待后续实验使用。之后制备Eu掺杂的HAP/Arg纳米粉,在量取Eu(NO3)3溶液与Ca(NO3)2溶液前设计 Eu3+与 Ca2+为某一固定的摩尔比,而(Ca2++Eu3+)与P摩尔比为1.67。制备Tb掺杂HAP/Arg纳米颗粒的方法也与制备Eu掺杂HAP/Arg纳米颗粒的方法相同。
1.5 HAP纳米颗粒混悬液的制备
取制备的HAP纳米颗粒样品0.5 g,放入离心管,加去离子水20 mL,用超声波分散60 min (Ultrasonic Hemogenizer24710, USA) ,配置成浓度为25 mg/mL的混悬液。然后静置观察2 h,混悬液应保持无分层,呈乳状。将纳米颗粒混悬液置入50 mL 玻璃瓶,高压灭菌后备用。取1 mL纳米颗粒混悬液样本,超声处理8 min后,用于纳米颗粒粒度与分散状态的透射电镜观察。
2 结果与讨论
2.1 纯HAP纳米颗粒的形貌
对水热法合成的颗粒样品进行了 TEM观察,其结果如图1所示。由图1可以看出,单个颗粒长度在50~100 nm,直径为20~30 nm。但是,颗粒样品的团聚比较明显,二次颗粒的粒径为几百纳米甚至达到微米级,图片中几乎没有见到单独存在的纳米 HAP颗粒。对颗粒样品进行TEM选区电子衍射(SAED)分析(见图1(b)),其衍射图符合HAP晶面的排列结构。可见,用水热法合成的粉末为纳米级的HAP。
2.2 纯HAP纳米颗粒Zeta电位与pH值的关系
HAP纳米颗粒混悬液的Zeta电位与体系pH值的关系如图2所示。从图2可以看出,当体系pH值为4左右时,HAP纳米颗粒Zeta电位几乎为0,接近体系的等电点;随着体系pH值的升高,Zeta电位绝对值也相应增大,在pH值为7时,Zeta电位绝对值达到最大值23.1 mV;pH值继续增大时,Zeta电位的绝对值逐渐减小,但减小幅度趋于逐渐平缓。

图1 纳米HAP的TEM像及样品的选区SAED谱Fig. 1 TEM image(a) and SAED pattern(b) of nano HAP synthesized by hydrothemal method

图2 HAP颗粒的Zeta电位随pH的变化Fig. 2 Changes of pH value with Zeta potential of HAP particle
2.3 纯HAP纳米颗粒的粒度与pH值的关系
图3所示为纳米HAP的粒度随混悬液体系pH值的变化。

图3 HAP颗粒粒度随pH的变化Fig. 3 Changes of pH value on particle size of HAP
从图3可以看出,HAP纳米颗粒的粒度随混悬液体系pH值的增加呈先减小后增大的趋势。当体系的pH值由4增加到7时,HAP颗粒的粒度下降比较明显,从约470 nm下降到370 nm,pH值为7左右时达到最小值,表明颗粒的分散性逐渐增加;当pH值由7增加到11时,HAP颗粒的粒度逐渐增大,但趋势比较平缓,表明混悬液体系中 HAP的分散效果相对较好。结合pH值与Zeta电位的关系也可以得出HAP颗粒粒度的变化趋势受 Zeta电位的影响很大,HAP纳米颗粒在pH=7时,Zeta电位的绝对值最大,因而体系中的HAP颗粒的分散性最好;当pH值在4左右时,HAP纳米颗粒表面电位为零,HAP颗粒的粒径变得最大。这说明当纳米HAP溶胶处于一定的pH值时,HAP颗粒吸附了一定量的电荷形成扩散双电层,使得溶胶中颗粒之间产生静电排斥作用。然而,水热合成的纯 HAP纳米颗粒的分散性仍无法满足基因载体的要求,因此,本文作者研究精氨酸对HAP纳米颗粒进行修饰改性。
2.4 HAP/Arg颗粒的显微形貌
采用TEM观察了所制备的HAP/Arg颗粒晶体形貌、颗粒粒度及分散状态,其结果如图4所示。

图4 水热合成HAP颗粒的TEM像Fig. 4 TEM images of HAP crystal synthesized by hydrothermal method: (a) Without arginine; (b) With 0.01mol/L arginine;(c) With 0.05mol/L arginine; (d) With 0.10mol/L arginine
从图4(a)可以看出,没有加入精氨酸时,水热法合成的HAP颗粒呈短柱状与等轴状。短棒状颗粒长约60~180 nm,同一颗粒的横截面尺寸均匀,约为30~70 nm,短棒状颗粒的长径比约2~4;等轴状颗粒的直径约为30 nm。HAP为密排六方结构,从颗粒的形状也能反映出HAP生长成成棒状和针状颗粒的习性。加入0.01mol/L精氨酸后,合成的HAP较没添加精氨酸合成的 HAP在颗粒的大小上稍有减小,形貌上大致相当,团聚现象也比较明显(见图4(b))。而加入0.05mol/L精氨酸后,同样条件下合成的HAP颗粒也为纳米级,较没加精氨酸修饰的HAP颗粒尺寸明显减小,短棒状颗粒的长度缩短至 50~80 nm,各方向的尺寸差异变小,其外形不如HAP颗粒规则,但颗粒的分散性更好(见图 4(c))。当精氨酸的添加浓度增加至 0.10mol/L时,同条件下合成的HAP颗粒形貌如图4(c)所示,可以看出,HAP颗粒的尺寸有进一步减小的趋势,但是颗粒的大小和形状的均匀性有所降低。
单纯的HAP纳米颗粒在酸性环境下Zeta电位一般为正,可通过静电作用与荷负电的DNA结合,但结合能力较低;在体内外中性或弱碱性的转染环境下Zeta电位为负,难以结合DNA。在本研究中,pH=7.4时,HAP粉末的Zeta电位为(-23.1±3.5) mV,其与荷负电的DNA难以结合。本实验中选取精氨酸(分子结构如图5所示)作为修饰物,一方面是由于亲水性的精氨酸为碱性氨基酸,带有胍基基团—(CH2)3NHC(NH2)+,其等电点约10.76,高于水热合成实验中反应液的pH值,因此,在整个水热反应过程中精氨酸荷正电;另一方面,研究发现精氨酸和胍基官能团能有效穿透细胞膜[12],可在DNA转运到细胞内的过程中发挥非常重要的作用,暗示富精氨酸短肽可能会有效提高基因转染效率,但富精氨酸短肽介导基因转染的机理仍不太清楚,明显有别于受体介导的内吞机制[13]。

图5 精氨酸分子结构图Fig. 5 Structural formula of arginine
实验数据表明,水热合成时添加精氨酸能够明显改变HAP颗粒的形貌,且浓度增加时这种差别也会更明显,其主要原因是HAP晶体的生长过程会由于精氨酸的介入会产生变化。在合成HAP的过程中,由于精氨酸在结晶析出 HAP晶粒表面的吸附存在晶面的选择性,影响 HAP 晶粒长大的行为。热力学平衡条件下,HAP 晶体的择优生长方向为[001]。由HAP的晶体结构可知,[001]方向上存在P点的吸附位置,由于P点偶尔会出现Ca2+离子空缺而带有负电荷,在水热介质中精氨酸带正电性的胍基基团—(CH2)3NHC(NH2)+可与HAP(001)面裸露的负电性羟基(—OH)发生静电作用,导致精氨酸倾向于吸附在HAP的(001)面。因此,精氨酸的吸附更大程度地阻碍了溶液合成产物在HAP(001)面的析出,使得HAP各晶体方向的生长速度差异减小,晶体各方向的尺寸差异降低。
另一方面,精氨酸的—NH2基团能与HAP的各晶面以氢键的形式结合,研究表明,在没有精氨酸的溶液体系中,HAP晶体表面会被一层水膜包覆,然而,HAP晶体与精氨酸之间的相互作用力比HAP晶体与水分子之间的作用力更强,或者说HAP晶体与精氨酸的作用能更低。因此,精氨酸加入以后易于吸附在HAP颗粒样品表面,尤其是在HAP晶体的(001)面,水分子的吸附将被精氨酸所取代。当HAP各个晶面吸附了一定数量的精氨酸之后,晶体生长受阻,要求更多的HAP新颗粒形核析出,以降低溶液的过饱和度。形核密度的提高有利于获得更细小的 HAP颗粒。因此,当精氨酸的浓度增加到一定程度后,HAP各晶面都将吸附不同数量的精氨酸,可能导致图4(d)出现的颗粒在大小和形状的均匀性上的差异。
2.5 HAP/Arg颗粒的Zeta电位
对分别添加0.05mol/L和0.10mol/L精氨酸合成的HAP/Arg颗粒样品在pH值为7.4 时进行了Zeta电位测试,其结果如图6所示。由图6(a)可看出,在弱碱性的条件下(pH=7.4),添加0.05mol/L精氨酸合成的HAP/Arg纳米颗粒的Zeta电位为(30.9±8.2) mV,而未修饰的HAP纳米颗粒的Zeta电位为(-23.1±6.5)mV。表明合成HAP的过程中加入精氨酸使得颗粒表面的Zeta电位由常规HAP的负值变为HAP/Arg纳米颗粒的正值。添加0.10mol/L精氨酸合成的HAP/Arg纳米颗粒在 pH=7.4弱碱性的条件下的 Zeta电位为(32.1±8.1) mV,如图6(b)所示,虽然较添加0.05mol/L精氨酸合成的HAP/Arg颗粒的Zeta电位稍有增加,但增加不明显,可能是本实验条件下合成HAP的过程中,精氨酸的含量相对过于饱和。从以上测试结果得出水热法制备 HAP颗粒过程中引入一定量的精氨酸可使合成的 HAP/Arg颗粒表面的荷电状态发生根本性的改变,而且Zeta电位的绝对值更大,表明本实验制备的HAP/Arg颗粒具有较好的分散性,将更有利于HAP纳米颗粒与DNA的静电结合。这一变化是因为HAP/Arg颗粒表面吸附了精氨酸或精氨酸残留物。
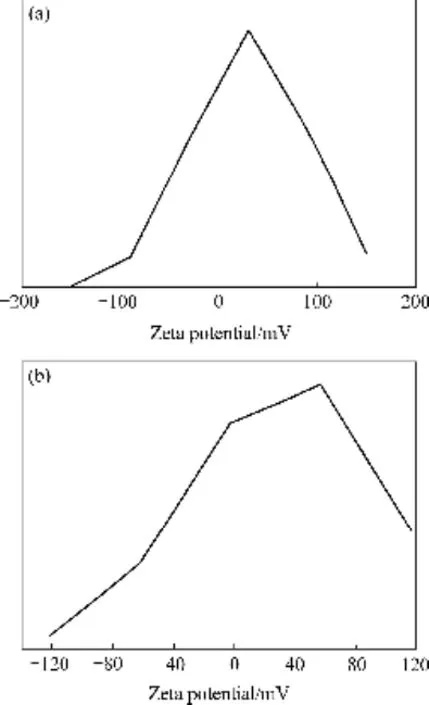
图6 HAP/Arg颗粒的Zeta电位图Fig. 6 Zeta potential of HAP/Arg particles: (a) With 0.05mol/L arginine; (b) With 0.10mol/L arginine
2.6 HAP/Arg颗粒物相组成
对水热法制备的HAP/Arg颗粒样品用XRD进行物相分析,扫描角度为25°~55°,速度采用2.4 (°)/min。图7所示为在反应过程中是否加入精氨酸样品的XRD谱。从图7可以看出,添加0.05mol/L精氨酸合成的HAP/Arg颗粒与没有加入添加精氨酸的 HAP颗粒样品的XRD谱基本相同,其特征峰尖锐,表明所制备颗粒的结晶程度较高。对主要特征峰所代表的晶面进行标定发现,所制备的HAP/Arg颗粒样品能很好地符合HAP国际标准粉末卡片JCPDs 9-432,从而表明所制备的样品是结晶完整的羟基磷灰石颗粒。但不同样品间主要特征峰相对强度存在细微差异,相对于(002)晶面的衍射峰,HAP/Arg颗粒样品的(300)晶面衍射峰的强度较HAP样品有所增大,而HAP/Arg颗粒样品的(002) 晶面的衍射峰与(211) 晶面的衍射峰强度的比值较HAP颗粒样品有所降低。
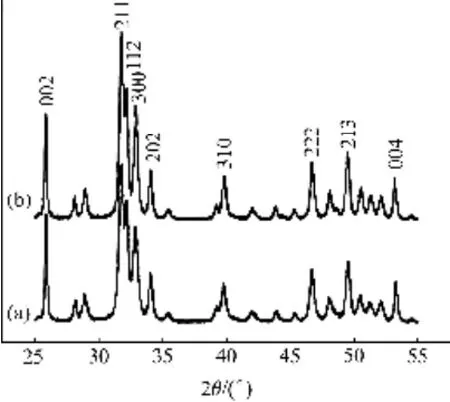
图7 160 ℃水热法合成的HAP颗粒的XRD谱Fig. 7 XRD patterns of HAP particles synthesized by hydrothermal method at 160 ℃: (a) Without arginine; (b) With arginine
2.7 HAP/Arg颗粒红外光谱特征
采用傅立叶红外光谱仪测试了所制备 HAP/Arg颗粒样品的红外光谱特征,结果如图8所示。从图8可以看出,没有加入精氨酸与加入精氨酸合成 HAP纳米颗粒具有相似的红外光谱,谱中主要峰的位置相同。较强的峰线出现在 565.25、604.21、1035.78、3441.75 cm-1等位置,并在1106.57、1420.30、1631.24 和3570.12 cm-1等位置出现强度较弱或位置较宽的峰线。理论上,磷酸根(PO43-)的4种振动方式对应峰的位置分别如下:ν1峰在960 cm-1附近,ν2峰位于470 至440 cm-1区域,ν3峰位于1190至976 cm-1区域,ν4峰位于在600至560 cm-1区域。因此,565.25、604.21 和1035.78 cm-1的强峰以及1106.57 cm-1的弱吸收峰是由HAP的PO43-产生的。晶格中水分子的特征峰出现在 3550~3200 cm-1区域,因此,3441.75和 3570.12 cm-1位置的峰是晶格水和磷灰石羟基(OH-)的反映。1631.24 cm-1处的特征峰是H2O的振动峰,表明颗粒样品表面吸附少量的水分。氨基(—NH2)的特征峰出现在 1400~1420 cm-1区间,1420.30 cm-1峰可能是少量来自于原料磷酸氢二氨的铵根(NH4+)、氨基酸的残留物吸附在HAP的反映。对于加入精氨酸的样品,该峰强度有所增强(见曲线b),说明存在精氨酸的残留物。通过红外光谱的比较,发现加入精氨酸合成HAP纳米粉末的吸收峰符合HAP的特征,表明在结构上与通常的HAP基本一致。

图8 水热合成HAPFTIR谱Fig. 8 FTIR spectra of HAP prepared by hydrothermal synthesis: (a) Without arginine; (b) With arginine
2.8 稀土掺杂HAP/Arg颗粒样品的物相
采用X射线衍射测试了掺杂不同含量Eu合成的HAP/Arg颗粒样品,结果如图9所示。由图9可以看出,不同Eu掺杂含量下HAP/Arg纳米颗粒样品具有基本相同的XRD谱,各样品的主要衍射峰峰形尖锐,其它衍射峰明显,且能与 HAP国际标准粉末卡片JCPDs 9-432中晶体衍射峰位置完全对应,但与EuPO4及Eu(OH)3标准图谱中的主要峰位对应不上,说明所合成的产物为单一物相的HAP。此外,随着Eu掺杂含量的增加,产物的主要衍射峰位向高角度方向逐渐有微小的移动,说明Eu的掺杂改变了HAP的晶格常数。Eu3+离子的半径(0.095 nm)与的 Ca2+离子的半径(0.099 nm)相当[14],在掺杂过程中较容易占据 Ca2+的位置,但由于其离子半径略小,Eu3+离子进入 Ca2+格位后使得HAP的晶格常数减小。根据X射线衍射的布拉格公式2dsinθ=nλ,在满足衍射的条件下,晶格常数的减小即衍射晶面间距的减小,将导致衍射角的增大,这与实验观察到的结果一致,而且进入Ca2+格位的Eu3+离子增多,HAP晶格常数的改变相对越大,因此衍射峰位移动也越明显。采用电感耦合原子发射光谱(ICP-AES)对Eu掺杂的HAP/Arg颗粒样品中Eu的浓度进行了检测,得到在较低的掺杂浓度(1%,摩尔分数)下,Eu3+在HAP晶格中的分凝系数可到0.9;然而掺杂浓度较高时(≥3%),Eu的分凝系数下降至0.75左右。
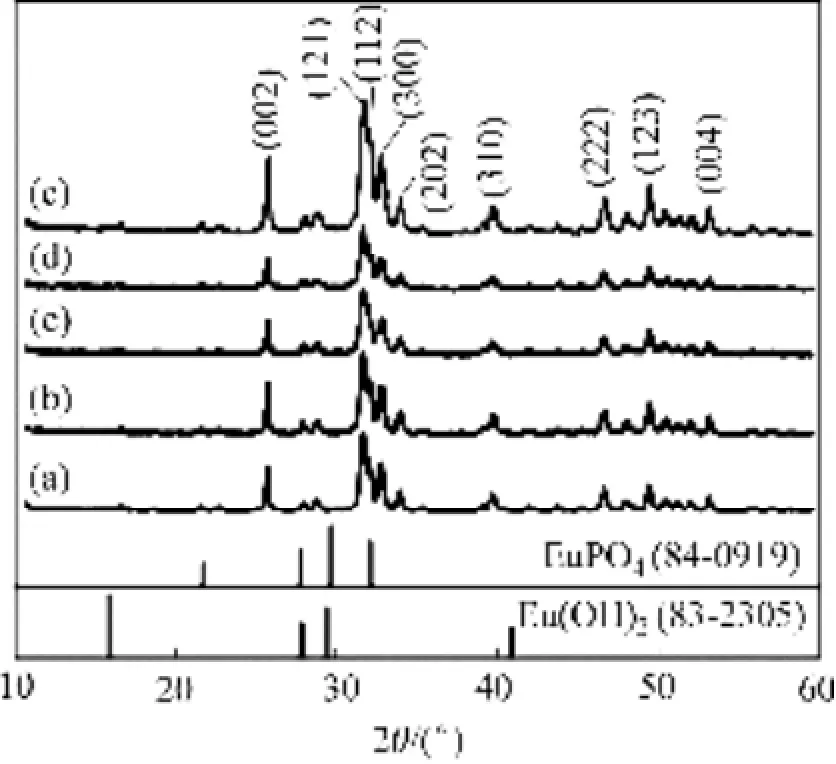
图9 不同Eu掺杂量HAP/Arg颗粒的XRD谱Fig. 9 XRD patterns of HAP/Arg particles with different dopants of Eu: (a) 0; (b) 1%; (c) 3%; (d) 5%; (e) 7.5%
由于图中不同浓度Eu掺杂的HAP/Arg颗粒各主要衍射峰的峰形比较尖锐,说明产物结晶程度较高,因此对各样品(002)和(300)晶面衍射峰的半高宽分别进行了测量,然后根据谢乐(Scherrer)公式计算了相应的晶粒尺寸:

式中:D(hkl)为计算得到的晶粒尺寸(nm);k为宽化常数(取0.9);λ为X射线的波长(为0.154056 nm);β为衍射峰的半高宽(FWHM,rad)。经过计算得出Eu掺杂的 HAP/Arg纳米颗粒沿 c轴方向尺寸稍大于 100 nm,a轴方向尺寸约为33 nm,说明所得到的产物的仍为纳米级。
同样,对掺杂2% Tb合成的HAP/Arg颗粒样品也进行了XRD分析,结果如图10所示。从图10可以看出,样品的主要衍射峰尖锐明显,与HAP的国际标准粉末卡片 JCPDs 9-432中晶体衍射峰位置完全对应,且没有出现TbPO4与Tb(OH)3等其他物相的衍射峰,说明所合成的产物也为单一物相的HAP。
通过对2% Tb掺杂的颗粒衍射数据的计算分析,得到晶格常数a为0.9437 nm,c为0.6899 nm,而未掺杂的HAP/Arg纳米颗粒晶格常数a为0.9454 nm,c 为0.6903 nm。可见,稀土Tb3+离子掺杂也使HA的晶格常数减小。这是由于Tb3+离子的半径(0.092 nm)小于Ca2+的离子半径[15],Tb3+离子替换部分 Ca2+离子后使得HAP的晶格常数和晶胞体积减小。采用ICP-AES 对Tb的浓度进行了检测,得到Tb3+离子在HAP晶格中的分凝系数约为0.96,可见离子半径更小的Tb3+更易掺杂进入HAP晶格。

图10 Tb掺杂HAP/Arg颗粒的XRD谱Fig. 10 XRD patterns of HAP/Arg particles doped with Tb
2.9 稀土掺杂HAP/Arg颗粒样品红外光谱特征
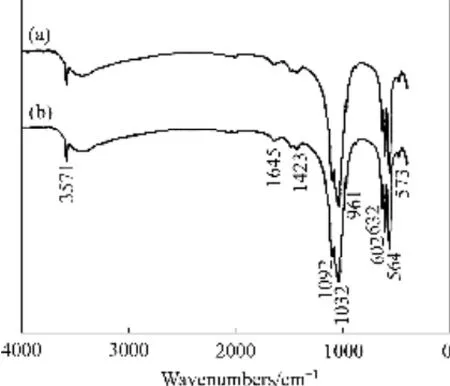
图11 HAP/Arg颗粒的FTIR谱Fig. 11 FTIR spectra of HAP/Arg particles: (a) Without Eu doping; (b) With Eu doping
对Eu掺杂HAP/Arg颗粒样品的FTIR光谱特征进行了测试与分析,结果如图11所示。从图11可以看出,Eu掺杂HAP/Arg颗粒与未掺杂HAP/Arg颗粒样品的红外光谱特征基本一致,谱线中主要峰位相同。其中,564和 602 cm-1属于 PO43-的 ν4吸收峰、573 cm-1为PO43-的ν2吸收峰,961 cm-1为PO43-的ν1吸收峰,1032和1092 cm-1为PO43-的ν3吸收峰,1432 cm-1处的特征峰是H2O的振动峰,3571 cm-1位置的峰是晶格水和磷灰石羟基(OH-)的反映。此外,1645 cm-1可能是CO32-的吸收峰[16]。图9所示为HAP/Arg纳米颗粒中掺杂 Eu未带来特征峰的明显位移以及其他吸收峰等变化,说明掺杂Eu对HAP/Arg纳米颗粒的结构无影响。
同样,对Tb掺杂的HAP/Arg颗粒样品也进行了FTIR测试,结果如图12所示。与Eu掺杂HAP/Arg颗粒样品的 FTIR光谱特征相同,Tb掺杂也未给HAP/Arg颗粒的结构带来影响。
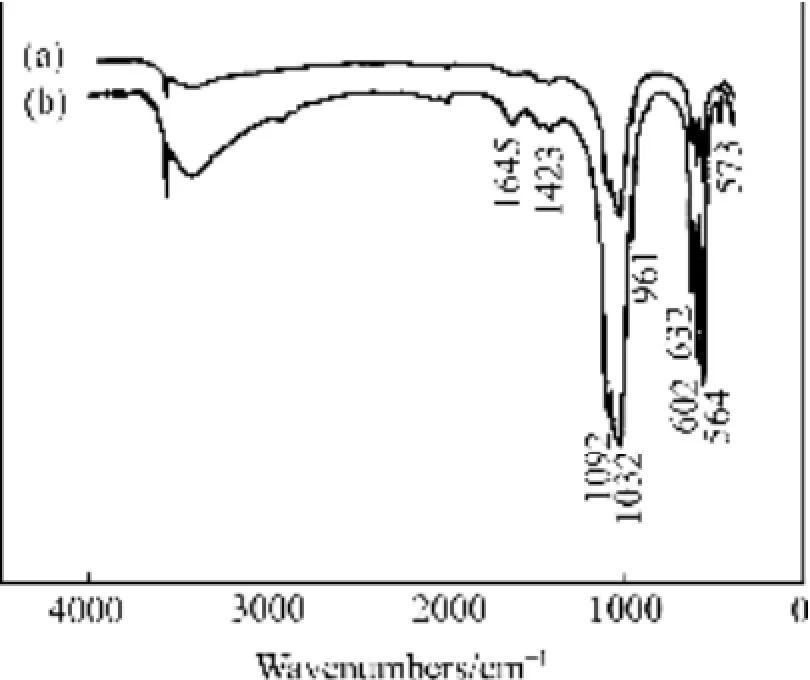
图12 HAP/Arg颗粒的FTIR谱Fig. 12 FTIR spectra of HAP/Arg particles: (a) Without Tb doping; (b) With Tb doping
2.10 稀土掺杂HAP/Arg粉末的TEM观察
图13所示为160 ℃下水热反应3 h制得的稀土掺杂的HAP/Arg颗粒的TEM像。由图13(a)可以看出,Eu掺杂HAP/Arg颗粒以短柱状为主,也有少量等轴状颗粒,短柱状颗粒大小比较均匀,长度约为100 nm左右,长径比 3~4,表明为纳米级的颗粒,这也与采用 XRD数据计算的结果比较接近。而 Tb掺杂的HAP/Arg颗粒也主要呈短柱状,并含有少量等轴状颗粒,颗粒大小与Eu掺杂HAP/Arg纳米颗粒相近,如图13(b)所示。由于Eu与Tb同属于镧系元素,具有相似的物理与化学性质,因此,在水热法合成HAP/Arg纳米颗粒过程中这两种元素的掺杂对颗粒生长的影响比较接近。
2.11 稀土掺杂HAP/Arg颗粒的荧光特性

图13 稀土掺杂HAP/Arg颗粒的TEM像Fig. 13 TEM images of HAP/Arg particles doped with Eu(a)and Tb(b)
对不同量Eu掺杂HAP/Arg颗粒的荧光光谱进行了测试与分析,结果如图14所示。图14(a)所示为Eu掺杂HAP/Arg纳米颗粒的激发光谱,监测波长为612 nm,可以看出在361.2、381.0及394.4 nm有3个特征激发峰,其中,最强的激发峰位于394.4 nm附近。HAP/Arg纳米颗粒中Eu的掺杂浓度增加,激发峰的强度也随之增大,未掺杂Eu的HAP/Arg纳米颗粒样品没有出现相应的激发吸收。图14(b)所示为Eu掺杂HAP/Arg纳米颗粒在波长为394 nm的激发光激发下的发射光谱。可以看出,未掺杂Eu的HAP/Arg纳米颗粒样品没有发射特征峰,而掺杂Eu的HAP/Arg纳米颗粒样品分别在580~600 nm及605~625 nm波段范围内出现发射特征峰,峰位在588.8和612.6 nm,分别对应于Eu3+的5D0-7F1与5D0-7F2跃迁。随掺杂量的增加,Eu3+的发射特征峰的强度也相应地增加,表明Eu3+掺杂进入了HAP晶格。
此外,612.6 nm的发光峰在激光共聚焦显微镜的扫描发射滤波器检测范围内,激发最强的394.4 nm的激发光接近可见光波段。然而,有一个激发波段在464.8 nm附近,接近于激光共聚焦显微镜的激发光波长(488 nm)。因此,在这个激发波长下可在可见光范围内观察到活细胞。

图14 Eu掺杂HAP/Arg颗粒的荧光光谱Fig. 14 Excitation(a) and emission(b) spectra of HAP/Arg particles sample with different dopants of Eu
图15所示为Tb掺杂HAP/Arg纳米颗粒的光致发光谱,激发光波长为272 nm。从图可以看出,未掺杂Tb的HAP/Arg纳米颗粒不具有特征发光峰,而掺杂2%(摩尔分数)Tb的HAP/Arg纳米颗粒在488.6、542.4、583.4和619.0 nm附近出现光致发光峰,分别对应于Tb3+离子的5D4-7F6、5D4-7F5、5D4-7F4和5D4-7F3跃迁[17]。其中,542.4nm处的发光峰具有最大的发光强度,在绿光范围内,可以用于观察HAP/Arg纳米颗粒对细胞的各种作用及其在细胞中的分布等。由此可见,Tb3+离子可以掺入HAP/Arg纳米颗粒中,实现HAP/Arg纳米颗粒的光致发光。
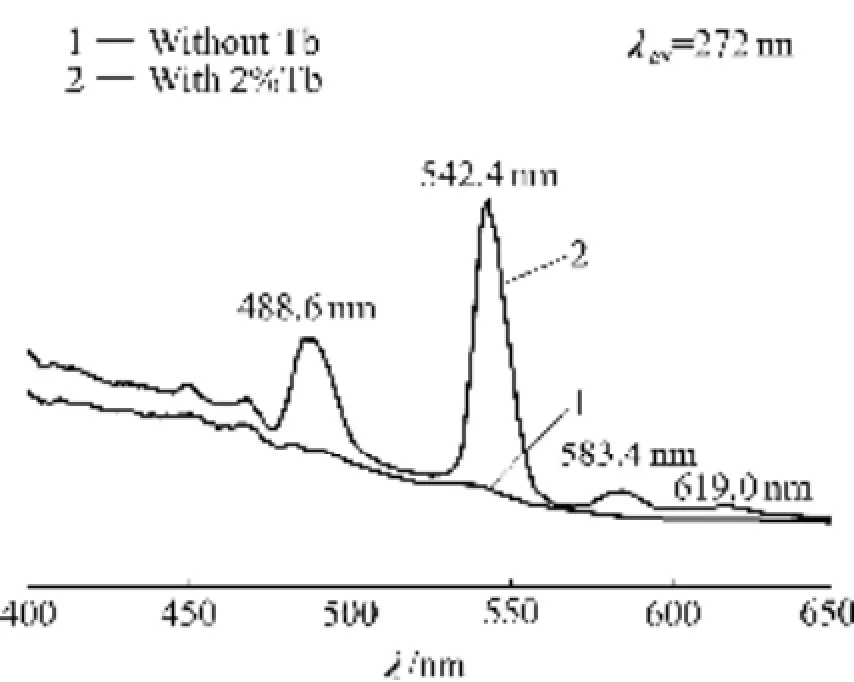
图15 Tb掺杂HAP/Arg颗粒的荧光光谱Fig. 15 Emission spectra of HAP/Arg particles sample with different dopants of Tb
3 结论
1) 制备了结晶程度比较高、晶形完整的HAP纳米颗粒,通过精氨酸表面的修饰改变了HAP纳米颗粒的表面Zeta电位,达到(30.9±8.2) mV,为提高HAP纳米颗粒作为基因转染载体的效能奠定了基础;研究发现精氨酸的加入在一定程度上抑制了 HAP的生长速率。在同样合成条件下,加入精氨酸修饰的 HAP短棒状颗粒的长度为50~80 nm,小于没加精氨酸修饰HAP纳米颗粒的60~180 nm,且长径比也有所减小,外形不如HAP纳米颗粒规则。
2) 少量稀土离子 Eu或 Tb掺杂不影响合成的HAP/Arg纳米颗粒产物的结构,均为单一的HAP物相,所合成产物的粒径也为纳米级。Eu掺杂的HAP/Arg纳米颗粒在580~601 nm和601~625 nm两个可见光波段范围内有发射特征峰,而 Tb掺杂的HAP/Arg纳米颗粒主要荧光特征发射峰位于488.6 nm 与542.4 nm。
3) 经稀土金属铽/铕掺杂的精氨酸表面修饰的HAP纳米颗粒可作为基因转染载体被运用。
REFERENCES
[1]BROWN W E, CHOW L C. A new calcium phosphate,water-setting cement[J]. American Ceramic Society, 1986, 4: 352-379.
[2] ZHU W, WANG D, XIONG J, LIU J, YOU W, HUANG J,DUAN L, CHEN J, ZENG Y. Study on clinical applicationof nano-hydroxyapatite bone in bone defect repair[J]. Artificial Cells Nanomedicine and Biotechnology, 2015, 43(6): 361-365.
[3] NORDSTROM T, SENKAS A, ERIKSSON S, PONTYNEN N,NORDSTROM E, LINDQVIST Ch. Generation of a new protein purification matrix by loading ceramic hydroxyapatite with metal ions-demonstration with poly-histidine tagged green fluorescent protein[J]. J Biotechnol, 1999, 69(2/3): 125-133.
[4]LEGEROS R Z. Properties of osteoconductive biomaterials: calcium phosphates[J]. Clinical Orthopaedics and Related Research, 2002, 395: 81-98.
[5]AOKI H, KUTSUNO T. An in vivo study on the reaction of hydroxyapatite-sol injected into blood[J]. Journal of Materials Science: Materials in Medicine, 2000, 11(2): 67-72.
[6]LIU Z S, TANG S L, AI Z L. Effects of hydroxyapatite nanoparticles on proliferation and apoptosis of human hepatoma BEL-7402 cells[J]. World Journal of Gastroenterology, 2003,9(9): 1968-1971.
[7]臧丽格. 不同羟基磷灰石纳米颗粒基因载体性能的比较研究[D]. 长沙: 中南大学, 2008. ZANG Li-ge. Comparison of the vector capacity of different modified hydroxyapatite nanoparticles as gene vectors[D]. Changsha: Central South University, 2008.
[8] DO T N, LEE W H, LOO C Y, ZAVGORODNIY A V,ROHANIZADEH R. Hydroxyapatite nanoparticles as vectors for gene delivery[J]. Therapeutic Delivery, 2012, 3(5): 623-632.
[9]毛岳峰. 基于壳聚糖修饰纳米羟基磷灰石基因载体的转染率研究[D]. 长沙: 中南大学, 2009. MAO Yue-feng. Study on the transfection efficiency of chitosan modified nano hydroxyapatite as gene transfer carrier[D]. Changsha: Central South University, 2009.
[10] ZUO G, WAN Y, ZHANG Y. Preparation and characterization of a novel laminated magnetic hydroxyapatite for application on gene delivery[J]. Materials Letters, 2012, 68: 225-227.
[11] 李 燕, 阳 俊, 刘桂英, 张 欣. 基因治疗药物输递系统的研究现状及发展趋势[J]. 生物化学与生物物理进展, 2013,44(10): 998-1007. LI Yan, YANG Jun, LIU Gui-ying, ZHANG Xin. Research situation and development trend of gene therapy for drug delivery system[J]. Progress in Biochemistry and Biophysics,2013, 44(10): 998-1007.
[12] 陶显东, 钟 镭, 薛 磊, 赵学维. 载顺铂的纳米羟基磷灰石对 A549肺腺癌细胞体外胀亡的影响[J]. 组织工程与重建外科杂志, 2012, 8(1): 14-17. TAO Xian-dong, ZHONG Lei, XUE Lei, ZHAO Xue-wei. Necrosis of the lung cancer cell line A549 treated with hydroxyapatite nanoparticles carried cisplatin in vitro[J]. Journal of Tissue Engineering and Reconstructive Surgery, 2012, 8(1): 14-17.
[13] 李湘南, 陈晓明, 彭志明, 李世普. 纳米羟基磷灰石/壳聚糖载药微球的制备及性能[J]. 中南大学学报(自然科学版), 2011,42(5): 1232-1237. LI Xiang-nan, CHEN Xiao-ming, PENG Zhi-ming, LI Shi-pu. Preparation and performance of drug-loaded nano-hydroxyapatite/ chitosan microspheres[J]. Journal of Central South University (Science and Technology), 2011, 42(5): 1232-1237.
[14] LIN H Y, FANG Y Ch, HUANG X R, CHU Sh Y. Luminescence and site symmetry studies of new red phosphors (Ca,Ba)3(VO4)2:Eu3+under blue excitation[J]. Journal of the American Ceramic Journal of the American Ceramic Society, 2010, 93(1): 138-141.
[15] 张若桦. 稀土元素化学[M]. 天津: 天津科学技术出版社,1987: 1-2. ZHANG Ruo-hua. Rare earth element chemistry[M]. Tianjin: Tianjin Science and Technology Press, 1987: 1-2.
[16] TERNANE R, PANCZER G, COHEN-ADAD M Th,GOUTAUDIER C, BOULON G, KBIR-ARIGUIB N,TRABELSI-AYEDI M. Relationships between structural and luminescence properties in Eu3+-doped new calcium borohydroxyapatite[J]. Optical Materials, 2001, 16: 291-300.
[17] RAMBABU U, AMALNERKAR D P, KALE B B,BUDDHUDU S. Optical properties of LnPO4:Tb3+(Ln=Y La and Gd) powder phosphors[J]. Materials Chemistry and Physics,2001, 70(1): 1-6.
(编辑 龙怀中)
Preparation and characteristics of functional nano-hydroxyapatite
ZHAO Yan-zhong1, 2, 3, YANG Min1, ZHANG Hai-bin2, ZHU Jun2, ZHOU Ke-chao2
(1. The Third Xiangya Hospital, Central South University, Changsha 410013, China;2. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China;3. Research Center for Medical Material and Instruments, Central South University, Changsha 410013, China)
Nano-hydroxyapatite(HAP) was prepared by a hydrothermal method with calcium nitrate, diammonium hydrogen phosphate as raw material, and to study its characteristics for morphology modification of arginine-functionalized and doped with rare earth, such as Eu3+or Tb3+. The crystallization, grain size and dispersibility of the sample HAP were analyzed and discussed. The results show that the surface Zeta potential of arginine-functionalized HAP is changed, and the growth rate of HAP is inhibited to a certain extent during the synthesis. The structure of HAP/Arg is not affected during the synthesis doped by a small amount of rare earth ions, such as Eu3+or Tb3+. All these samples have single phase of HAP with good dispersibility. The synthesized HAP is also nano-sized level. Nano-hydroxyapatite with arginine functionalized and rare earth doped, such as Eu3+or Tb3+, is suitable for the application of gene delivery as a gene carrier.
hydroxyapatite; hydrothermal synthesis; arginine modification; rare-earth doping
Project(2013SK2024) supported by the Key Project in Social Development Pillar Program of Hunan Province, China; Project(20130162120094) supported by Special Research Fund for the Doctoral Program of Higher Education of China; Project supported by Open Project of State Key Laboratory of Power Metallurgy of Central South University, China
date: 2015-11-10; Accepted date: 2016-05-25
ZHAO Yan-zhong; Tel: +86-0731-88618669; E-mail: yanzhongzhao@163.com
R318
A
1004-0609(2016)-06-1235-11
湖南省科技计划重点项目(2013SK2024);教育部博士学科点基金资助项目(20130162120094);中南大学粉末冶金国家重点实验室开放课题
2015-11-10;
2016-05-25
赵颜忠,副教授,博士;电话:0731-88618669;E-mail: yanzhongzhao@163.com
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
体育科技文献通报(2022年4期)2022-10-21
军事文摘(2022年12期)2022-07-13
少儿科技(2022年2期)2022-03-05
中国听力语言康复科学杂志(2021年6期)2021-12-21
食品安全导刊(2021年20期)2021-11-28
猪业科学(2018年7期)2018-01-22
中外医疗(2015年11期)2016-01-04
中国当代医药(2015年30期)2015-03-01
数理化学习·教育理论版(2013年9期)2013-12-27
