
烃类化合物燃烧热的QSPR研究
2016-08-08 08:56刘焯戎杨玉良殷海青祁正兴
贵州师范大学学报(自然科学版) 2016年1期
张 朋,刘焯戎,杨玉良,殷海青,祁正兴
(青海民族大学 化学化工学院,青海 西宁 810007)
烃类化合物燃烧热的QSPR研究
张朋,刘焯戎,杨玉良,殷海青,祁正兴*
(青海民族大学 化学化工学院,青海 西宁810007)
摘要:通过ChemOffice 8.0计算了32种烃类化合物的10个量子化学参数,并采用SPSS13.0统计学软件建立了烃类化合物燃烧热的最佳预测方程。结果表明:烃类化合物的燃烧热与分子碳氢含量及不饱和度密切相关,该方程对烃类化合物的燃烧热有很好的预测效果。
关键词:量子化学;回归方程;烃类化合物;燃烧热
0引言
标准燃烧热(简称燃烧热)是有机化合物的一个重要的化学热力学参数[1]。有机烃类物质的加氢、脱氢及燃烧反应等均要利用燃烧热来计算化学反应热,继而为质能联算以及反应器和燃烧炉的设计提供依据。燃烧热是衡量有机化合物火灾危险程度的重要特征量[2]。由于现代化学工业的发展,新的有机化合物不断涌现,对其燃烧热进行逐个的检测存在一定的困难,所以建立一个直观、准确的有机物燃烧热的预测模型,对于其物理化学参数的丰富是很有必要性的[2,3]。在科学研究和工程应用中数据的缺乏和不足,提供合理性的基础理论指导。
定量结构-性质相关性(QSPR)的量子化学研究方法是近年来化学、安全环境科学等学科研究中的一个重要的科学前沿领域[4,5]。其研究的基本依据是根据化合物的性质和结构二者之间具有的相关性,即以描述化合物结构的方法与其性质建立相关数学模型,并由此模型来预测未知化合物的一些性质参数。也有大量的QSPR研究,根据分子结构预测化合物性能的模型和方法发展,特别是在预测燃烧热研究方面,以不同物系的有机物理化性质及活性、燃烧热的QSPR研究,也已建立了不同的相应预测方法和模型。文章以ChemOffice 8.0对32种烃类化合物进行结构参数的计算,结合多元逐步回归分析建立QSPR模型,探讨根据分子结构预测有机物燃烧热的途径以及烃类化合物的燃烧热与分子碳氢含量及不饱和度的关系,并通过采用QSPR的分析方法对烃类化合物燃烧热进行回归模型的建立。
1数据来源及建模方法
1.1数据来源
燃烧热Q(单位KJ·mol-1)数据主要来自于文献[6]。
采用ChemOffice 8.0对32种烃类化合物分子进行了结构参数的计算。具体的计算过程如下:
1) 通过ChemDraw ultra 8.0绘出烃类化合物的分子结构,然后对分子结构进行整理;
2) 启动Chem3D ultra 8.0并对分子结构进行能量最小化与结构最优处理;
3) 通过Chem3D ultra 8.0中的半理论半经验法计算出32种烃类化合物的最高占据轨道能(EHOMO,单位eV),最低占据轨道能(ELUMO,单位eV),总能量(TE,单位eV),排斥能(NRE,单位eV),电子能(ElcE,单位eV),生成热(HF,单位eV)和偶极矩(μ)7个量子化学参数。
1.2建模方法
采用SPSS 13.0统计软件进行多元逐步回归分析来建立QSPR模型。以上述32种烃类化合物的7个量子化学参数及其含碳个数N、不饱和度m及含氢个数h共10个参数为逐步回归的自变量,以32种烃类化合物的燃烧热为因变量进行多元逐步回归分析建立QSPR模型。
2结果与讨论
2.1回归模型的建立
按照相对偏差小,相关系数大的原则,采用SPSS 13.0统计学软件,将32种烃类化合物的燃烧热Q与10种量子化学参数(部分具体数据见表1)进行逐步回归分析得到烃类化合物燃烧热的多元回归方程:
Q=-0.935+0.275N+0.241m+0.223h-0.079EHOMO-0.072ELUMO+0.066ElcE
(n=32,R=1.000,R2=1.000,S=0.025,F=22 994.4,P=0.000)
其中,n表示样本中化合物的数目,R表示相关系数,S表示标准偏差,F表示Fisher检验值,P表示显著性水平。
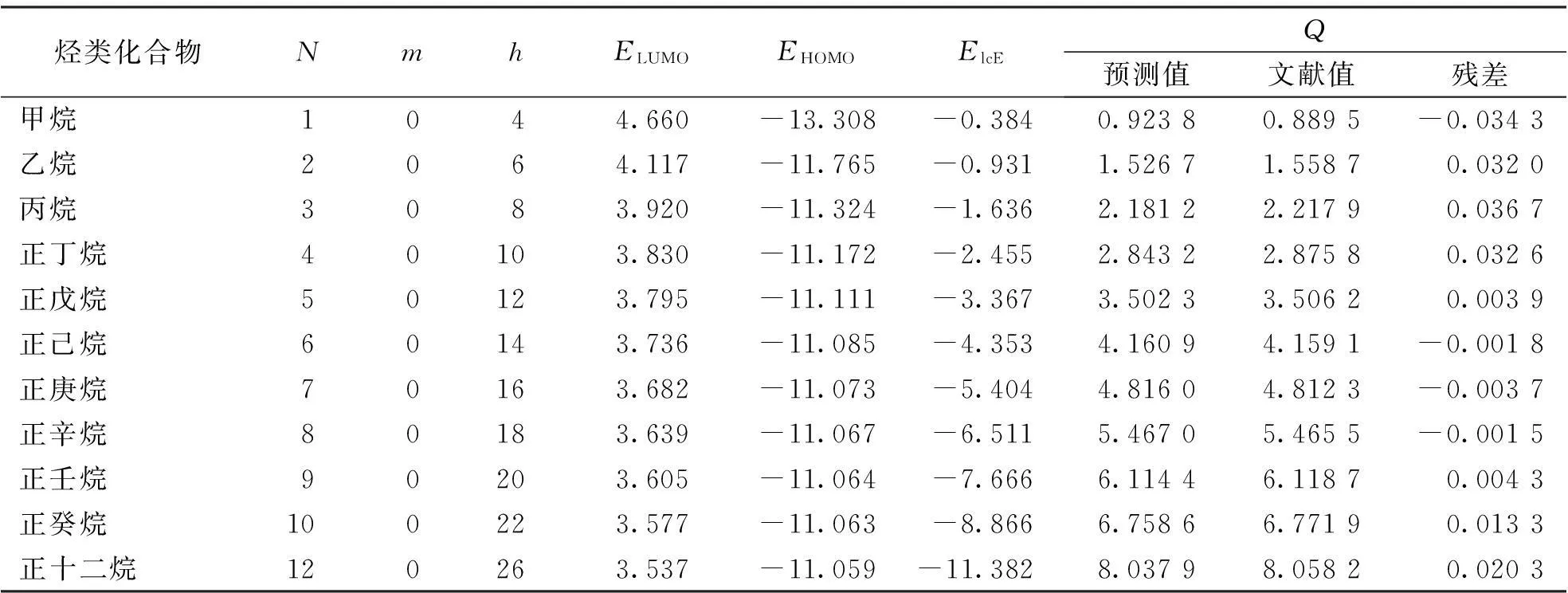
表1 烃类化合物的结构参数及燃烧热的预测值、文献值、残差
续表1
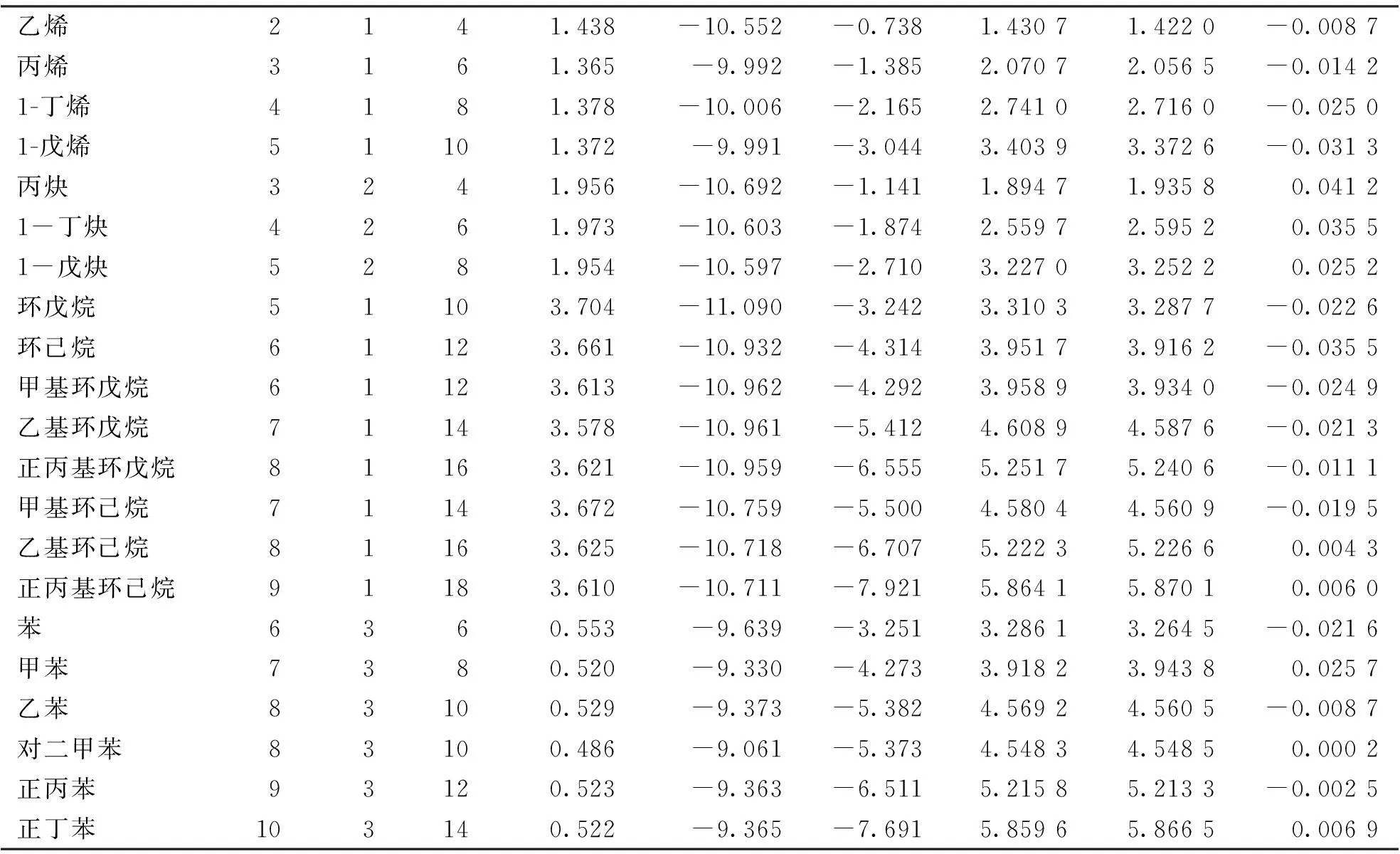
乙烯2141.438-10.552-0.7381.43071.4220-0.0087丙烯3161.365-9.992-1.3852.07072.0565-0.01421-丁烯4181.378-10.006-2.1652.74102.7160-0.02501-戊烯51101.372-9.991-3.0443.40393.3726-0.0313丙炔3241.956-10.692-1.1411.89471.93580.04121-丁炔4261.973-10.603-1.8742.55972.59520.03551-戊炔5281.954-10.597-2.7103.22703.25220.0252环戊烷51103.704-11.090-3.2423.31033.2877-0.0226环己烷61123.661-10.932-4.3143.95173.9162-0.0355甲基环戊烷61123.613-10.962-4.2923.95893.9340-0.0249乙基环戊烷71143.578-10.961-5.4124.60894.5876-0.0213正丙基环戊烷81163.621-10.959-6.5555.25175.2406-0.0111甲基环己烷71143.672-10.759-5.5004.58044.5609-0.0195乙基环己烷81163.625-10.718-6.7075.22235.22660.0043正丙基环己烷91183.610-10.711-7.9215.86415.87010.0060苯6360.553-9.639-3.2513.28613.2645-0.0216甲苯7380.520-9.330-4.2733.91823.94380.0257乙苯83100.529-9.373-5.3824.56924.5605-0.0087对二甲苯83100.486-9.061-5.3734.54834.54850.0002正丙苯93120.523-9.363-6.5115.21585.2133-0.0025正丁苯103140.522-9.365-7.6915.85965.86650.0069
2.2回归模型的评价
2.2.1内部评价
从所建立的回归模型可以看出,燃烧热的预测值和文献值的拟合曲线的相关系数R可达到1.000,并且其R2=1.000表明该模型具有很好的预测能力及稳健性;Fisher检验值较大,显著性概率P=0.000,并且由表2所示的回归方程中各系数的t检验值和显著性概率也可知模型的稳定可靠。同时,由表1中可以得知文献值和根据回归方程得到的预测值十分接近,这说明所建立的回归方程的预测效果较好,可以通过此回归方程很好的预测烃类化合物的燃烧热。
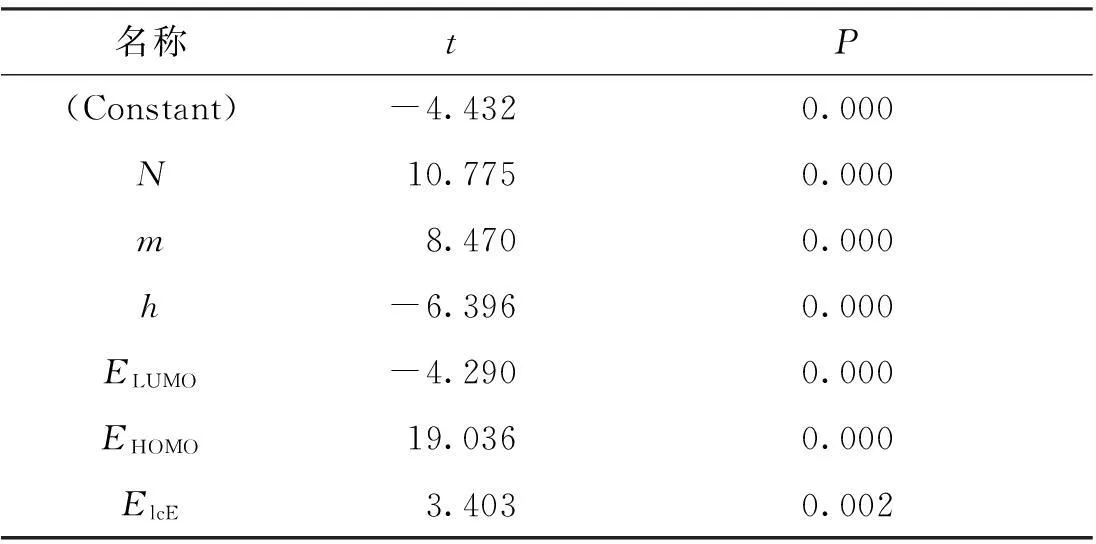
表2 回归方程中回归系数的t检验值与显著性概率
2.2.2外部评价
根据建立的回归方程和表1我们可以得到残差正态分布图(图1)、烃类化合物燃烧热的残差散点图(图2)和文献值与预测值的拟合曲线(图3)。
由图1可以看出烃类化合物燃烧热的残差值以零点位置为中心呈正态分布,符合统计学规律,很好的反映了回归模型预测的准确性。根据图2烃类化合物燃烧热的残差的散点分布可以看出残差的分布比较均匀,且图3显示回归模型的预测值与文献值非常接近,说明所建立的回归模型的预测能力较好。

图1 烃类化合物燃烧热残差的正态分布Fig.1 The normal distribution of residuals of the heat of combustion of hydrocarbon compounds
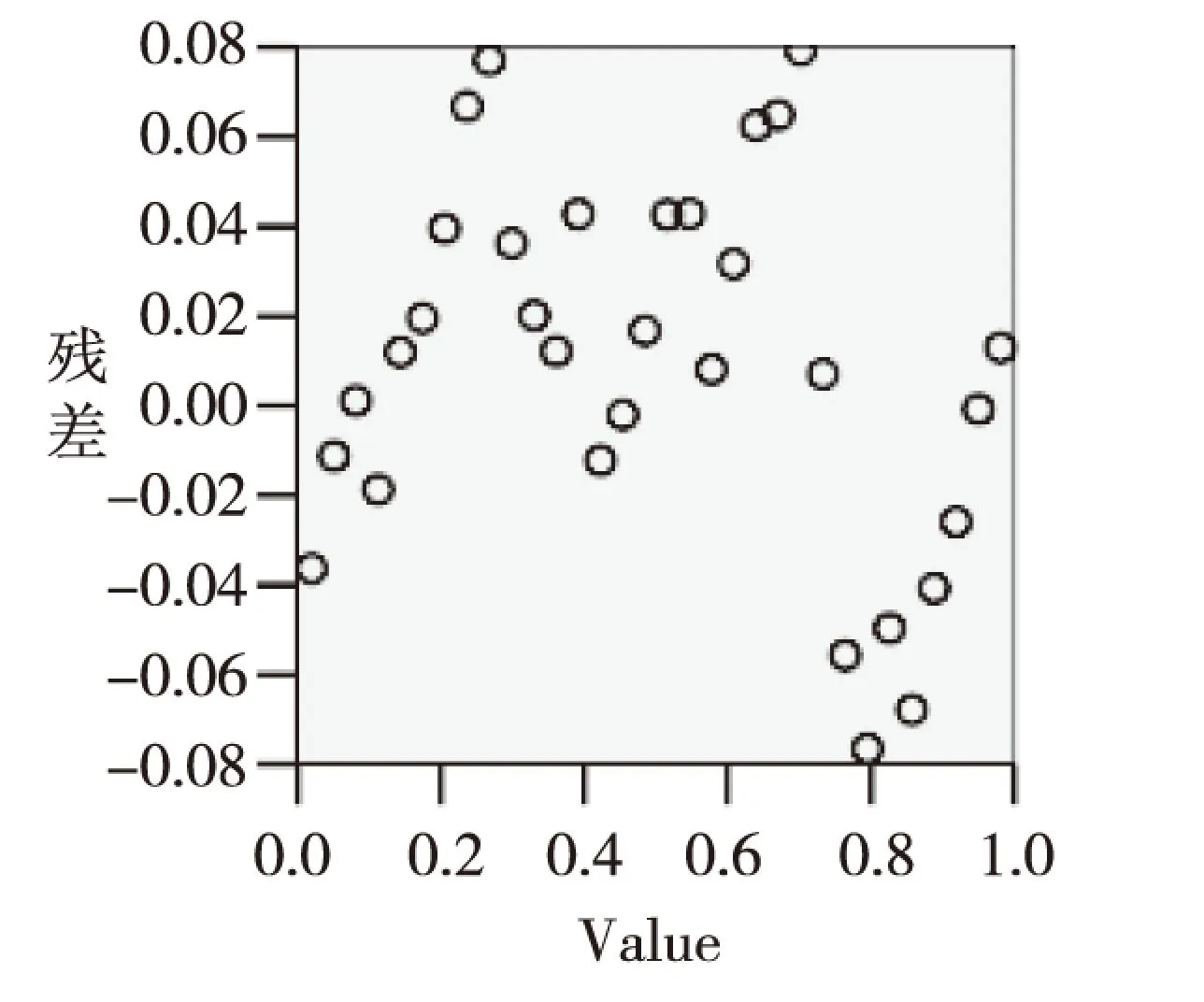
图2 烃类化合物燃烧热的残差散点图Fig.2 The residuals scatter plot of the heat of combustion of hydrocarbon compounds
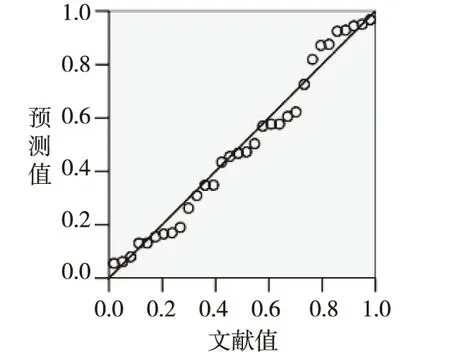
图3 烃类化合物燃烧热的预测值和文献值的关系Fig.3 Relationship of predictive value and literature values of the heat of combustion of hydrocarbon compounds
3结论
对烃类化合物燃烧热的QSPR研究可知,影响烃类化合物燃烧热的主要因素是有机化合物分子的含碳个数(N)、含氢个数(h)及其不饱和度(m);次要因素是最高占据轨道能(EHOMO)、最低占据轨道能(ELUMO)和电子能(ElcE)。我们可以通过所建立的模型来粗略的预测不同烃类化合物的燃烧热。
利用QSPR的分析方法可以建立直观、稳定的模型来预测烃类化合物的燃烧热,为我们更好的丰富烃类化合物的理化参数提供理论依据。当然,由于样本的有限性和计算数据获取的途径的差异性,个别分子的预测值偏差较大,这需要我们在以后的工作中进一步加强对烃类化合物燃烧热的研究。
参考文献:
[1] 彭昌军.链烷烃标准燃烧热的拓扑计算法[J].天然气化工:化学与化工,1996,21(6):53-56.
[2] 曹洪印,蒋军成,潘勇.应用原子类型AI指数预测烃类燃烧热[J].工业安全与环保,2010,36(12):8-9.
[3] 孙立卿,周长会,李珊珊,等.量子化学参数对含氮杂环化合物的活(毒)性研究[J].山西大同大学学报(自然科学版),2014,30(3):44-46.
[4] 周从艺,聂长明,文松年,等.扩展的邻接矩阵指数AI及对烷烃的QSPR/QSAR研究[J].分析科学学报,2007,23(2):137-142.
[5] 张金生,汪荣凯,王光彦.NO2大π键量子化学研究[J].贵州师范大学学报(自然科学版),2011,29(4):76-79.
[6] 彭津.烃类燃烧热的简易推算[J].消防科学与技术,1999,4(2):9-11.
文章编号:1004—5570(2016)01-0061-04
收稿日期:2015-03-30
基金项目:国家自然科学基金资助项目(21361021)
作者简介:张朋(1990-),男,硕士研究生,研究方向:有机化学,E-mail:qhmdzp_2013@163.com. *通讯作者:祁正兴(1962-),男,教授,硕士生导师,研究方向:化学计量学,E-mail:qi.zx@163.com.
中图分类号:O626
文献标识码:A
Quantitative structure-property relationship research on the heat of combustion of hydrocarbon compounds
ZHANG Peng, LIU Chaorong, YANG Yuliang, YIN Haiqing, QI Zhengxing*
(School of Chemistry and Chemical Engineering, Qinghai University for Nationalities, Xining,Qinghai 810007, China)
Abstract:This paper calculated ten structure parameters of 32 hydrocarbon compounds by using ChemOffice 8.0. And by using SPSS 13.0 statistical software, it established the best equation to predict the heat of combustion of hydrocarbon compounds. The results showed that the heat of combustion of hydrocarbon compounds was closely related to the hydrocarbon content of the molecule and the degree of unsaturation. This equation had a very good effect in predicting the heat of combustion of hydrocarbon compounds.
Key words:quantum chemistry; regression equation; hydrocarbon compounds; heat of combustion
猜你喜欢
科学与财富(2021年36期)2021-05-10
中学生数理化·高一版(2021年2期)2021-03-19
中学生数理化·高一版(2021年2期)2021-03-19
中学生数理化·高一版(2021年2期)2021-03-19
中学生数理化(高中版.高二数学)(2019年6期)2019-06-24
中学生数理化·高一版(2018年2期)2018-04-04
科技创新与应用(2017年14期)2017-05-19
中学生数理化·高二版(2017年1期)2017-04-18
中学生数理化(高中版.高二数学)(2017年1期)2017-04-16
中学生数理化·高三版(2016年9期)2016-05-14
