
6,7LiD的分子势能函数、力常数和光谱常数
2016-07-26 05:30:54李如松方国明宋启星刘文杰姬国勋牛莉博
核化学与放射化学 2016年1期
关键词:分子结构
李如松,方国明,宋启星,刘文杰,何 彬,姬国勋,许 鹏,牛莉博
1.第二炮兵工程大学,陕西 西安 710025;2.中国人民解放军96373部队,青海 西宁 810011
6,7LiD的分子势能函数、力常数和光谱常数
李如松1, 2,方国明2,宋启星2,刘文杰2,何彬1,姬国勋1,许鹏1,牛莉博1
1.第二炮兵工程大学,陕西 西安710025;2.中国人民解放军96373部队,青海 西宁810011
摘要:为了提供6,7LiD与H2O之间潮解机制研究所需的解析势能函数,采用B3LYP/6-311G(2df,pd)方法对6LiD、7LiD分子进行了几何结构优化,并在相同理论水平上进行单点能扫描计算,通过用核运动效应修正Born-Oppenheimer(B-O)近似下的电子能量。利用Murrell-Sorbie势能函数,得到了体现同位素原子质量差异的同位素双原子分子势能函数,并计算得到了力常数和光谱数据,其值与实验数据基本一致。
关键词:分子结构;势能函数;电子状态;反应静力学
6LiD化学性质非常活泼,可与空气中的水蒸气发生反应,在水中这种反应更为剧烈。粉末状的6LiD在潮湿的空气中可以自燃,有时甚至发生粉尘爆炸。此外还易与其它有机溶剂反应,如乙醇、乙醚等。这些材料极易与水反应,会使产品潮解、变质,品位下降,并且反应生成的氢(氘、氚)氧化锂是一种强碱,具有很强的腐蚀作用。为了保证质量,产品表面均涂有保护层。保护层轻微脱落者,必须按规定的工艺进行修补。为了防止材料的潮解,在LiH/LiD部件的表面涂上一层薄的防潮物质,但这只能使潮解减弱,原因是防潮物质的透湿系数不为零,从而导致在长期贮存过程中该材料潮解和品位降低。
中国工程物理研究院正在从实验方面研究氢化锂保护膜的老化规律与机制,认为在潮湿的空气中,最初和湿气反应生成LiOH层,然后LiOH再和其它的氢化锂缓慢反应得到Li2O和H2,最后在LiH表面会形成LiOH或Li2O层。而在相对湿度大于15%空气中暴露时,倾向于生成LiOH·H2O[1-2]。李赣等[3]进而研究了氘化锂粉末与O2、CO2和水汽的反应动力学,结果表明,氘化锂与O2、CO2的反应很微弱,而与水汽的反应速率相对要大的多。Dinh和Plummer等[4-6]采用程序升温方法研究了LiOH的分解过程以及水汽存在下LiH的腐蚀速率,分析了LiH的氧化腐蚀机理。徐梅等[7]分别运用二次组态相互作用QCISD(T)/6-311++G(3df,2pd)方法和电子相关单双耦合簇CCSD(T)/6-311++G**研究了氢化锂(LiH)分子基态的结构与势能函数。罗德礼等[1]基于相对论有效原子实势(RECP)和密度泛函(B3LYP/SDD)方法,优化得到了LiH的Murrell-Sorbie分析势能函数。谢安东等[8]利用SAC/SAC-CI方法,使用D95(d)、6-311G**及cc-PVTZ等基组,对LiH分子的基态(X1Σ+)、第一激发态(A1Σ+)及第二简并激发态(B1Π)的平衡结构和谐振频率进行了优化计算,利用SAC的GSUM(group sum of operators)方法对基态(X1Σ+)、SAC-CI的GSUM方法对激发态(A1Σ+和B1Π)进行单点能扫描计算,用正规方程组拟合函数,得到了相应电子态的完整势能函数,所得结果均与实验符合较好。雷洁红等[9-10]研究了LiX(X=H、D、T)与水的反应,计算了反应体系最低势能面上各驻点的构型参数、振动频率和能量,并计算了反应的速率常数。同时采用密度泛函理论方法研究了高温高压条件下LiD的热力学性质与温度和压强之间的关系[11-12]。
然而,目前几乎没有从微观角度揭示6,7LiD与H2O之间的潮解机制。因此,本工作拟利用量子化学方法计算6,7LiD分子的势能函数、力常数和光谱常数,为下一步6,7LiD与H2O之间反应的反应通道、势能曲面和H同位素效应研究提供重要的理论依据。
1计算方法
在Born-Oppenheimer(B-O)近似下由于忽略了核的运动效应,使得6LiD和7LiD分子与7LiH分子势能函数、力常数、离解能等具有不可区分性[13-15]。本工作提出一种电子能量修正方法,通过计算6LiD和7LiD分子与7LiH分子不同的核运动能量,修正在B-O近似下计算得到的电子能量,以获得体现氢、锂同位素质量差异的各分子基态的势能值,同时,通过优化计算确定6LiD和7LiD分子基态的稳定构型和平衡几何参数,确定其正确的离解极限。
采用Gaussian09软件,使用二次组态相互作用(quadratic configuration interaction,QCISD)和密度泛函B3LYP方法,分别结合6-311G(df,pd)、6-311G(df,2pd)、6-311G(2df,pd)和6-311G(2df,2pd)基组对7LiD分子基态进行了几何优化,并通过理论计算,优选出最佳的方法与基组组合为B3LYP/6-311G(2df,pd)。使用该法与基组组合分别对6LiD和7LiD分子基态进行了几何优化及势能扫描计算,经过核动能修正,得到相应的势能值,然后用非线性最小二乘法拟合出Murrell-Sorbie函数的参数,并由此计算出各阶力常数和光谱常数。
2结果和讨论
2.16LiD和7LiD分子基态的结构参数
采用QCISD和B3LYP方法,分别结合6-311G(df,pd)、6-311G(df,2pd)、6-311G(2df,pd)和6-311G(2df,2pd)基组,对7LiD分子基态进行结构优化计算,由优化结果可知B3LYP/6-311G(2df,pd)为最佳方法与基组组合。利用优选出的方法与基组分别对6LiD和7LiD分子基态(X1Σ+)进行优化计算,结构优化结果列入表1,其相应的电子能量和核运动能列入表2。
同位素分子离解能(De)的计算如公式(1)[16-18]。
De=xEe(M)+yEe(N)-Ee(MxNy)+
(1)
其中:Ee,电子能量;En,核运动能量;x、y,分子中M原子和N原子的个数。
表16LiD和7LiD分子基态(X1Σ+)结构优化结果
Table 1Structural optimization results of6LiD and7LiD ground state (X1Σ+)

分子电子态r/nmDe/eVωe/cm-16LiDX1Σ+0.15922.4071077.327LiDX1Σ+0.15922.4101057.87
注:r为Li-D平衡原子间距,ωe为谐振频率
表26LiD和7LiD分子的电子能量和核运动能
Table 2Electron energies and nuclear motion energies of6LiD and7LiD molecules

分子Ee(7Li)/eVEe(6Li)/eVEe(H)/eVEe(D)/eVEe(T)/eVEe(M)/eVEn(M)/(K·Cal·mol-1)6LiD-203.876635-13.666678-220.0120483.0387LiD-203.883139-13.666678-220.0132463.012
2.26LiD和7LiD的分子势能函数与光谱性质
目前,分子势能函数没有考虑核运动效应,它仅仅是在B-O近似下的电子本征能量函数,此时近似地把电子运动看作不受核运动的影响,因此在B-O近似下,具有相同电子数和不同核质量的同位素分子势能函数不可区分。实际上,分子核运动客观存在,并且会影响电子运动,从而导致其势能函数与现行B-O近似下不同程度的差别。文献[19-20]提供了同位素分子H2与T2以及OH与OD的实测光谱常数,结果列入表3。ωe为谐振频率,χe为非谐振因子,Be和ae分别为刚性和非刚性转动因子。
表3同位素分子H2、T2、OH和OD的实测光谱常数[19-20]
Table 3Experimental spectrum constants for isotopic molecules H2, T2, OH and OD[19-20]
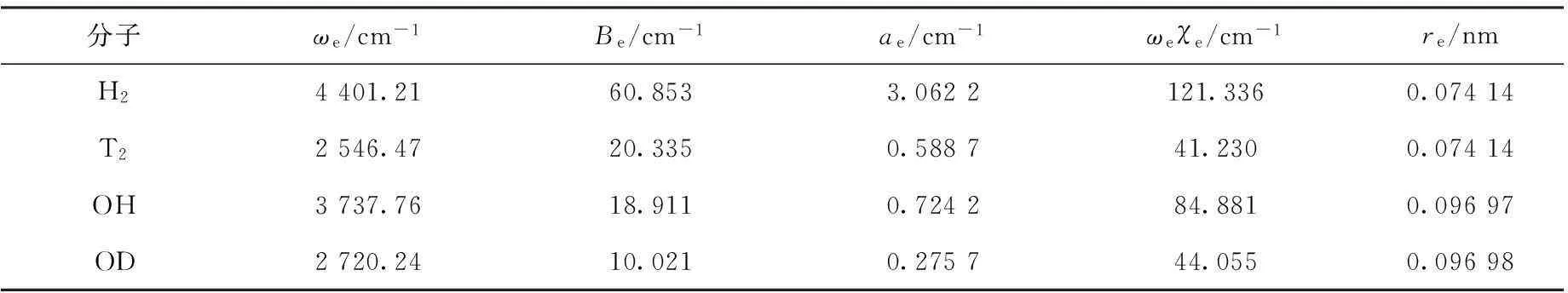
分子ωe/cm-1Be/cm-1ae/cm-1ωeχe/cm-1re/nmH24401.2160.8533.0622121.3360.07414T22546.4720.3350.588741.2300.07414OH3737.7618.9110.724284.8810.09697OD2720.2410.0210.275744.0550.09698
由表3可知,尽管同位素分子H2与T2以及OH与OD电子数相同,但由于核的质量不同,其光谱性质有所不同,且同位素效应很明显,所以B-O近似仅为一种有限的近似。现在的问题是如何修正B-O近似下的势能函数,使其进一步接近实际值以体现同位素分子势能函数的差异。现行的势能函数又称分子内势能函数,由于内坐标与核运动密切相关,因而与振动能Ev、约化质量μ和力常数k等相关。Ev、μ和k与ω、振动量子数(v)之间关系如式(2)、(3)[20]。式中:h为普朗克常数,c为光速。
(2)
(3)
所以,同位素分子的势能函数可以从Ev、ω和μ几个方面考虑进行校正。例如,7LiH(D,T)和6LiH(D,T)分子可用核的振动、转动和平动能修正B-O近似下的电子能量,以获得体现氢、氚同位素差异的势能值,并求得体现核运动效应的力常数。
为描述双原子分子的解析势能函数,到目前为止已经提出了大量的函数。其中,Murrell-Sorbie势能函数是迄今为止最好的解析势能函数形式之一,不仅在吸引支和排斥支均比H-H函数好,而且与RKR光谱数据非常相似。Murrell-Sorbie函数一般形式如式(4)。
(4)
式中:ρ=r-re,r和re分别为核间距和平衡核间距;De为离解能;ai为拟合的参数;De、ai均由拟合得到。由于多项式的顺序可以影响拟合的结构,所以对不同的分子,n的取值也不同。通过拟合工作,发现截止到9次方项可以得到很好的拟合结果。所以在拟合的过程中Murrell-Sorbie函数形式如式(5)。
(5)
因此,要获得分子的势能函数,首先要确定(5)式中的9个参数a1—a9和离解能De。这些量可以通过一系列单点能的计算,获得不同核间距下的势能值来拟合得到。为了获得体现同位素差异下的势能函数,本工作对每一个分子均计算了80多个不同核间距的电子能量和相应的核动能,用核动能修正电子能量,得到修正后的势能值,即:V=En+Ee。最后,用最小二乘法拟合得到势能函数参数,根据力常数与各个参数之间的关系式(6)—(8)[21-22],计算出各力常数,计算结果列入表4。
(6)
(7)
(8)
表46LiD和7LiD分子基态的Murrell-Sorbie势能函数参数与力常数
Table 4Murrell-Sorbie potential parameters and force constants of6LiD and7LiD ground states

分子a1/nm-1a2/nm-2a3/nm-31016f2/(J·nm-2)1015f3/(J·nm-3)1014f4/(J·nm-4)6LiD12.50-55.45172.72105.50-3595.0490488.117LiD12.51-55.37172.74106.53-3595.5390484.71
图1—2分别给出采用Murrell-Sorbie函数拟合的6LiD和7LiD分子基态势能函数,拟合数据与理论计算值之间的相关系数为0.999。

○——理论计算值,曲线是拟合值图1 B3LYP/6-311G(2df, pd) 水平下6LiD分子基态势能值与Murrell-Sorbie函数拟合势能曲线Fig.1 Potential values and Murrell-Sorbie fitting potential curve of 6LiD ground state at B3LYP/6-311G(2df, pd) level
在B-O近似下,分子势能函数是电子本征能量函数,近似地把电子运动看作不受核运动的影响,因此同一种元素的同位素分子的势能函数均相同。实际上,核运动肯定要影响电子运动,例如,H2与D2、 DT以及OH与OD的实测光谱数据有所不同,可见同位素效应明显,本工作采用文献[19,23-24]的方法,即B3LYP/6-311G(2df,pd)方法计算得到了6LiD和7LiD分子基态的平衡间距、离解能及谐振频率,7LiD计算值与实验值很接近。修正了同位素双原子分子的势能函数,所得光谱数据也与实验值(6LiD分子不存在可获的实验数据)符合得很好。

○——理论计算值,曲线是拟合值图2 B3LYP/6-311G(2df, pd)水平下7LiD分子基态势能值与Murrell-Sorbie函数拟合势能曲线Fig.2 Potential values and Murrell-Sorbie fitting potential curve of 7LiD ground state at B3LYP/6-311G(2df, pd) level
根据计算得到的二阶、三阶和四阶力常数,利用文献[25]的方法及公式(9)、(10)[7,20]计算得到6LiD和7LiD分子基态的光谱常数(式(11)、(12)),并给出了相应的实验数据,结果列入表5。
表56LiD和7LiD分子基态的光谱常数
Table 5Spectrum data of6LiD and7LiD ground states

分子ωe/cm-1Be/cm-1ae/cm-1ωeχe/cm-16LiD1092.3994.43970.087515.1567LiD1077.9234.28120.080714.2301055.121)4.23381)0.091981)13.2281)
注:1) 实验值
(9)
(10)
(11)
(12)
还可以利用双原子分子的同位素效应,根据已有的7LiD分子基态的光谱实验数据,获得6LiD分子基态的光谱数据。如果两个同位素分子中一个的光谱常数右上角标以i,由于同位素效应,则两个同位素的分子光谱常数间关系如式(13)—(16)[19-20]。
(13)
(14)

(15)
(16)
这些关系式可用于同位素分子之间光谱常数的相互推导,计算结果列入表6。对比表5和表6可知,本工作计算6LiD和7LiD的各项光谱数据与实验值和由实验值反演的相关数据符合得很好,这表明该理论方法可行。
由表5还可知,计算结果与实验数据都符合很好,所以用B3LYP/6-311G(2df,pd)方法计算出6LiD分子的ωe、Be、ae、ωeχe数据比较精确。所研究的6LiD和7LiD分子基态的势能函数同位素效应不如文献[9-10,24]所报道的H同位素双原子分子的明显,其原因可能是,H同位素之间的质量差别很大,当考虑核运动效应后,氢同位素分子的势能函数将有一定的差异,特别是在平衡位置附近更为明显,而所研究同位素分子之间的质量差别不是十分明显,故同位素效应小。由图1—2可知,经过修正后的两个分子的势能函数曲线十分接近,因此小分子同位素效应比大分子的同位素效应要大。
表67LiD光谱实验数据推导的同位素分子光谱常数
Table 6Spectrum data of isotopic molecules deduced from experimental spectrum data of7LiD

分子μ/μiωe/cm-1Be/cm-1ae/cm-1ωeχe/cm-16LiD0.57141073.5824.3830.095013.5337LiD0.05261054.2374.2260.090013.050
3结论
采用B3LYP/6-311G(2df,pd)方法对6LiD和7LiD分子进行了几何结构优化,并在相同理论水平上进行单点能扫描计算,通过用核运动效应修正B-O近似下的电子能量。利用Murrell-Sorbie势能函数,得到了体现同位素原子质量差异的同位素双原子分子势能函数,并进一步计算得到了力常数和光谱数据,其与实验得到的结果基本一致,可用于进一步研究分子的反应动力学特征,也为同位素分子的研究提供一定的理论依据。
参考文献:
[1]罗德礼,蒙大桥,朱正和.LiH,LiO和LiOH的分析势能函数与分子反应动力学[J].物理学报,2003,52(10):2438-2442.
[2]朱志艳,朱正和,张莉,等.T+OD体系的同位素交换反应动力学[J].物理学报,2011,60(12):123102.
[3]李赣,陆光达,敬文勇,等.氘化锂粉末与O2,CO2和水汽的反应动力学研究[J].核化学与放射化学,2004,26(2):99-102.
[4]Dinh L N, Cecala C M, Leckey J H, et al. The effects of moisture on LiD single-crystals studied by temperature-programmed decomposition[J]. J Nucl Mater, 2001, 295: 193-204.
[5]Dinh L N, Grant D M, Schildbach M A, et al. Kinetic measurement and prediction of the hydrogen outgassing from the polycrystalline LiH/Li2O/LiOH system[J]. J Nucl Mater, 2005, 347: 31-43.
[6]Plummer G M, Herbst E, de Lucia F C. Submillimeter spectra and molecular constants of6LiH,7LiH,6LiD, and7LiD[J]. J Chem Phys, 1984, 81: 4893-4897.
[7]徐梅,令狐荣锋,汪荣凯,等.LiH基态分子(X1Σ+)的结构与势能函数[J].贵州师范大学学报(自然科学版),2007,25(1):56-58.
[8]谢安东,施德恒,朱遵略,等.LiH分子X1Σ+、A1Σ+、B1Π态的势能函数[J].化学物理学报,2005,18(5):776-780.
[9]雷洁红,段浩,邢丕峰,等. LiX(X=H,D,T)与水的反应机理和动力学研究[J].原子能科学技术,2011,45(10):1165-1169.
[10]雷洁红,邢丕峰,唐永建,等.密度泛函理论研究LiX(X=H,D,T) 体系的热力学性质[J].强激光与粒子束,2010,22(10):2308-2312.
[11]段涛,雷洁红,唐永建.高温高压下LiD热力学性质的密度泛函理论研究[J].原子能科学技术,2011,45(4):394-397.
[12]雷洁红,邢丕峰,唐永建,等.高压下LiD热力学性质的密度泛函理论研究[M]∥中国核科学技术进展报告.北京:原子能出版社,2009:16-20.
[13]钟正坤,张莉,孙颖,等.氢-水同位素交换反应热力学理论研究[J].原子能科学技术,2015,49(2):250-254.
[14]耿振铎,樊晓伟,张岩松.XY(H,Li,Na)分子基态的结构与势能函数[J].物理学报,2006,55(5):2175-2179.
[15]樊晓伟,耿振铎,张岩松.OH分子基态(X2Π)的结构与势能函数[J].物理学报,2005,54(12):5614-5617.
[16]朱正和.原子分子反应静力学[M].北京:科学出版社,1996.
[17]江文世,吴开映.LiH,BeH和BH基态分子结构与势能函数[J].四川师范大学学报(自然科学报),2005,28(4):469-471.
[18]李权.钚化合物分子及分子离子的势能函数和分子反应动力学[D].成都:四川大学,2001.
[19]罗文浪,阮文,张莉,等.氢同位素氚水T2O(X1A1)的解析势能函数[J].物理学报,2008,57(8):4833-4839.
[20]朱正和,俞华根.分子结构与分子势能函数[M].北京:科学出版社,1997.
[21]谢安东,朱正和.BF分子X1Σ+,A1Π和B1Σ+电子态的势能函数[J].化学学报,2005,63(23):2126-2130.
[22]许永强,高晓明,张为俊.CuC、CuN分子基态的结构与分析势能函数[J].物理化学学报,2007,23(7):1075-1079.
[23]张莉,钟正坤,朱志艳,等.氢同位素双原子分子的解析势能函数[J].化学物理学报,2003,16(6):455-458.
[24]王蓉,张莉,蒋刚,等.基于核运动效应下的H同位素双原子分子的解析势能函数[J].原子核物理评论,2009,26(2):150-153.
[25]杜泉,王玲,谌晓洪,等.BeH,H2和BeH2的分子结构和势能函数[J].物理学报,2009,58(1):178-184.
收稿日期:2015-10-10;
修订日期:2015-12-05
基金项目:国家自然科学基金资助项目(51401237,51271198,11474358);第二炮兵工程大学基金项目(2014QNJJ018,YX2012cxpy06)
作者简介:李如松(1983—),男,安徽五河人,博士,讲师,从事核材料科学计算研究
中图分类号:O641.12
文献标志码:A
文章编号:0253-9950(2016)01-0013-06
doi:10.7538/hhx.2016.38.01.0013
Potential Functions, Force Constants and Spectrum Constants for6, 7LiD Molecules
LI Ru-song1,2, FANG Guo-ming2, SONG Qi-xing2, LIU Wen-jie2, HE Bin1, JI Guo-xun1, XU Peng1, NIU Li-bo1
1.The Second Artillery Engineering University, Xi’an 710025, China;2.Army 96373 of the Chinese People’s Liberation Army, Xining 810011, China
Abstract:To provide analytical potential functions for possible mechanisms for6,7LiD reactions with H2O, geometrical optimization of6LiD and7LiD molecules with B3LYP/6-311G (2df, pd) method was adopted. Then single-point energies at the same level were scanned, and electronic energies using nuclear motion effect corrected Born-Oppenheimer approximation were obtained. Diatomic molecule potentials manifestation of isotopic atomic mass difference was acquired using Murrell-Sorbie function. Force constants and spectrum constants by calculation are consistent basically with available experimental data.
Key words:molecular structure; potential function; electronic state; reaction statics
猜你喜欢
中学化学(2024年5期)2024-07-08 09:24:57
中学化学(2024年2期)2024-06-17 04:01:47
小猕猴智力画刊(2021年11期)2021-11-28 13:10:02
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
钻井液与完井液(2018年5期)2018-02-13 01:07:44
中学化学(2016年10期)2017-01-07 08:37:06
新乡学院学报(2016年6期)2016-12-01 05:21:37
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:40
化学教学(2015年6期)2015-09-15 02:50:02
中国石油大学学报(自然科学版)(2013年6期)2013-03-11 18:35:20
