
利用滑动窗口和KNN算法识别差异甲基化区域
2016-07-14 09:40李华兵
杭州电子科技大学学报(自然科学版) 2016年4期
李华兵,杨 昆
(杭州电子科技大学计算学院,浙江 杭州 310018)
利用滑动窗口和KNN算法识别差异甲基化区域
李华兵,杨昆
(杭州电子科技大学计算学院,浙江 杭州 310018)
摘要:针对现有差异甲基化区域DMRs识别方法中过度删除显著性弱的甲基化位点、DMRs长度受限以及不能直接处理多类的问题,提出了一种利用滑动窗口和KNN算法识别不同类别间DMRs的算法.算法先通过滑动窗口结合KNN分类器筛选候选区域,再根据误差率合并候选区域得到DMRs.真实数据上的实验表明,算法的分类性能、聚类指数明显优于对照算法,扩展了对照的Ong算法识别的DMRs长度,并能发现Ong算法未发现的DMRs.
关键词:差异甲基化区域;滑动窗口;KNN分类器;多类问题;聚类指数
0引言
DNA甲基化是指在DNA甲基转移酶(DNA methyltransferase,DNMTS)作用下,将甲基添加到碱基上,是一种重要的表观遗传修饰[1].不同条件下的生物学样本之间存在差异甲基化区域(Differentially methylated regions,DMRs),可能参与到基因表达的调控,进而影响基因功能.相关研究表明,相对于单个位点独立的识别方法,针对整个区域的识别方法更有生物学上的价值.DMRs识别与通常意义上的特征选择有显著区别:通常的特征选择往往假定特征间无关联性,然而DMRs是基因组上有位置关联的一个区域,而且长的DMRs更有生物价值.识别DMRs是当前研究领域中重要且新颖的研究问题,有别于通常的特征选择问题.目前,已有多个识别DMRs方法的提出,主要分3类:一类是通过分析计算一个区域中每个位点的差异来确定一个DMR,如bump hunting算法[2]和Slieker算法[3],这类算法缺点是过度删除了一些显著性弱的单个位点;第二类算法是通过相邻位点间的相关性,先聚类甲基化位点,然后估计每组聚类间差异性,如Ong算法[4],这类算法的不足之处是在识别DMRs之前区域长度受限;第三类算法是一种利用判别分析的DDA算法[5],这类算法针对两个类别的问题,不能直接应用于多类别问题.针对上述存在的问题,本文提出了一种利用滑动窗口和K-近邻(k-Nearest Neighbor,KNN)分类器的DMRs识别算法(Slide Windows KNN Algorithm,SWKA),并通过实验分析对比了算法的有效性和准确度.
1问题的描述和SWKA算法
1.1问题的描述
令p个位点n个样本的DNA甲基化数据看作一个矩阵M=(xij)n×p,其中p个位点看作p个特征,代表p个维度,每个行向量ti=(xi1,xi2,…,xip)可看作为模式识别中的一个模式(pattern),其中xij是芯片测出的样本ti位点j的甲基化水平值.不同类别样本中其DNA上存在甲基化程度明显差异的CpG位点,具有位置关联的差异甲基化位点构成DMRs.给定多个类别的DNA甲基化数据,识别基因组上连续的DMRs是本文的研究问题,本文主要针对Infinium 450 K甲基化数据开展工作.
1.2SWKA算法
SWKA算法是利用滑动窗口结合KNN分类器识别DMRs算法.算法详细流程如图1所示,首先划分各个染色体,探针不均匀的分布在染色体上,如图1(a)所示,其中圆圈表示位点,并设定窗口滑动步长k;利用滑窗方式将所要分析片段划分为位置关联的小片段(种子),如图1(b),其中实心和空心圆圈分别表示甲基化位点和非甲基化位点;再利用KNN分类器估计每个种子对样本的分类能力,选取分类误差率小于误差率阈值的种子为DMRs种子,如图1(c);在满足合并后分类误差率小于合并前的原则下,将有重叠的DMRs种子合并,得到候选DMRs,如图1(d);在相邻候选DMRs间距离小于一定长度和合并后的分类效果优于合并前的条件下对2个相邻候选DMRs进行合并,并对其进行显著性检验,最后得到满足P值条件的DMRs,如图1(e).获取本文算法源代码,发送主题为“获取利用滑动窗口和KNN分类器识别差异甲基化区域算法代码”的电子邮件到通信作者或者第一作者邮箱lucky_leehb@163.com.本文算法伪代码如下所述:
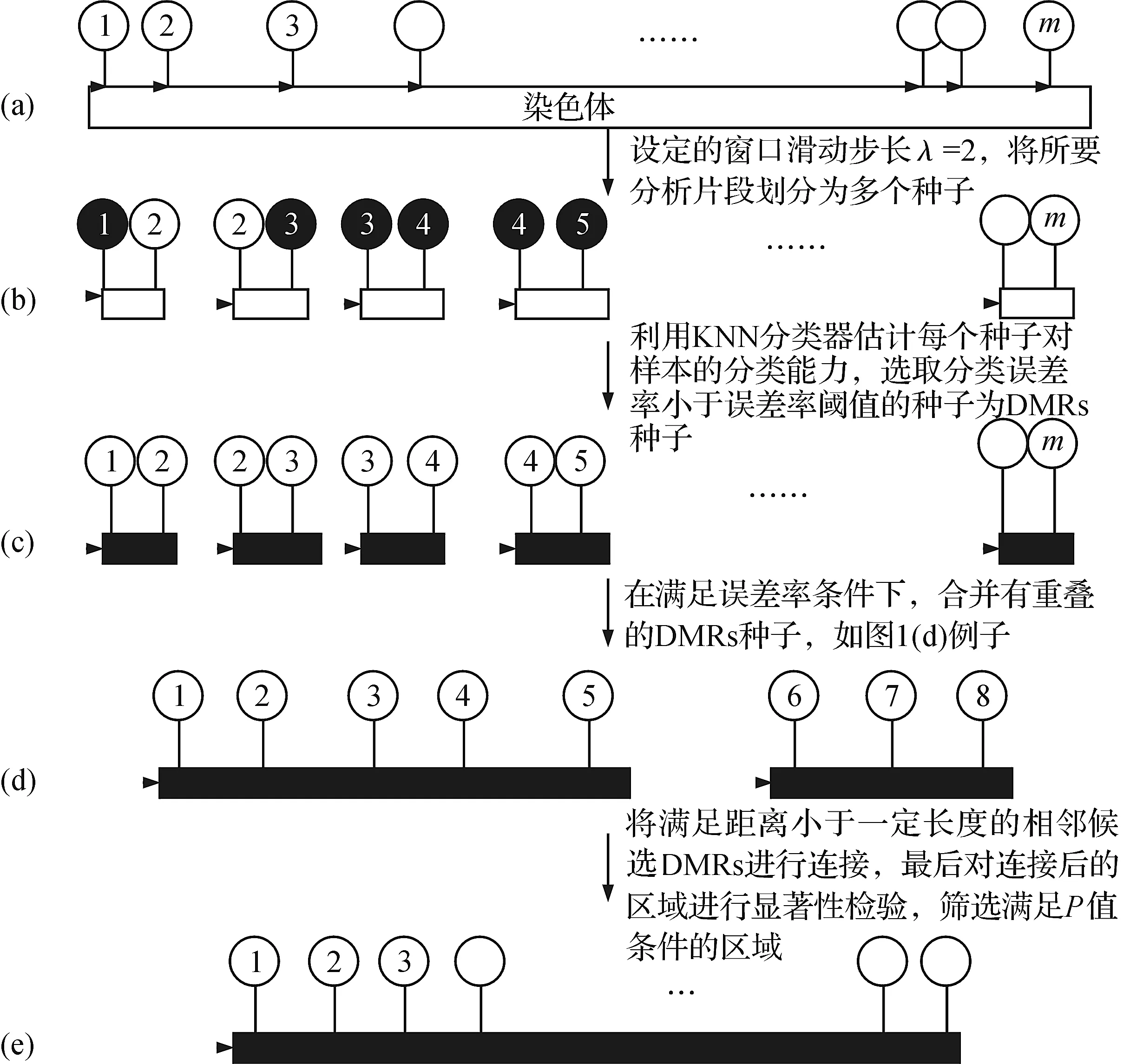
图1 识别DMRs的详细流程
2实验结果与分析
2.1实验环境和数据来源
本文实验在windows10 64-bit,Intel CPU i5 2.5 G四核、内存8.0 GB的PC上完成.
真实实验数据集(从GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)下载)是Infinium 450 K的4个年龄组甲基化数据集,来自GSE36064[6],GSE33233[7]和GSE30870[7],包含78个小孩样本、19个中年样本、新生儿和百岁以上老人样本各19个,每个样本包含485 577个位点.采用KNNimpute法[8]对每组缺失数据估计.实验中采用留一法交叉验证(LOOCV),其分类误差率阈值e为0.05,KNN分类器参数K为5,P阈值a为0.05,滑动窗口滑动步长k为2,相邻位点间距不大于1 000 bp.DDA和Ong算法是最新的识别DMRs方法,因为DDA法不能直接处理多类问题,所以本文采用Ong等人提出的DMRs识别算法作为实验对照方法.
2.2结果分析
本文采用参考聚类验证技术的Silhouette index[9]聚类指数和分类误差率为评价指标来考查所识别的DMRs.聚类指数是采用一个全局Silhouette index值GSu来作为聚类划分的有效评价指标,GS值越大表示聚类划分效果越好,计算公式如下,详细信息查阅文献[9].
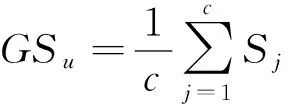
(1)
在对4个年龄组甲基化数据中,本文SWKA算法共识别出220个DMRs,使用Ong算法共识别出3 841个DMRs.本文识别出的220个DMRs中有161个区域不同于使用Ong算法计算出来的DMRs,两者重叠区域有59个.在重叠区域中有8个(13.56%)扩展了Ong算法识别的DMRs长度,11个(18.64%)与Ong识别的DMRs相同,22个(37.29%)与Ong识别的DMRs有重合,还有18个(30.51%)是包含于Ong发现的DMRs中,如图2所示.
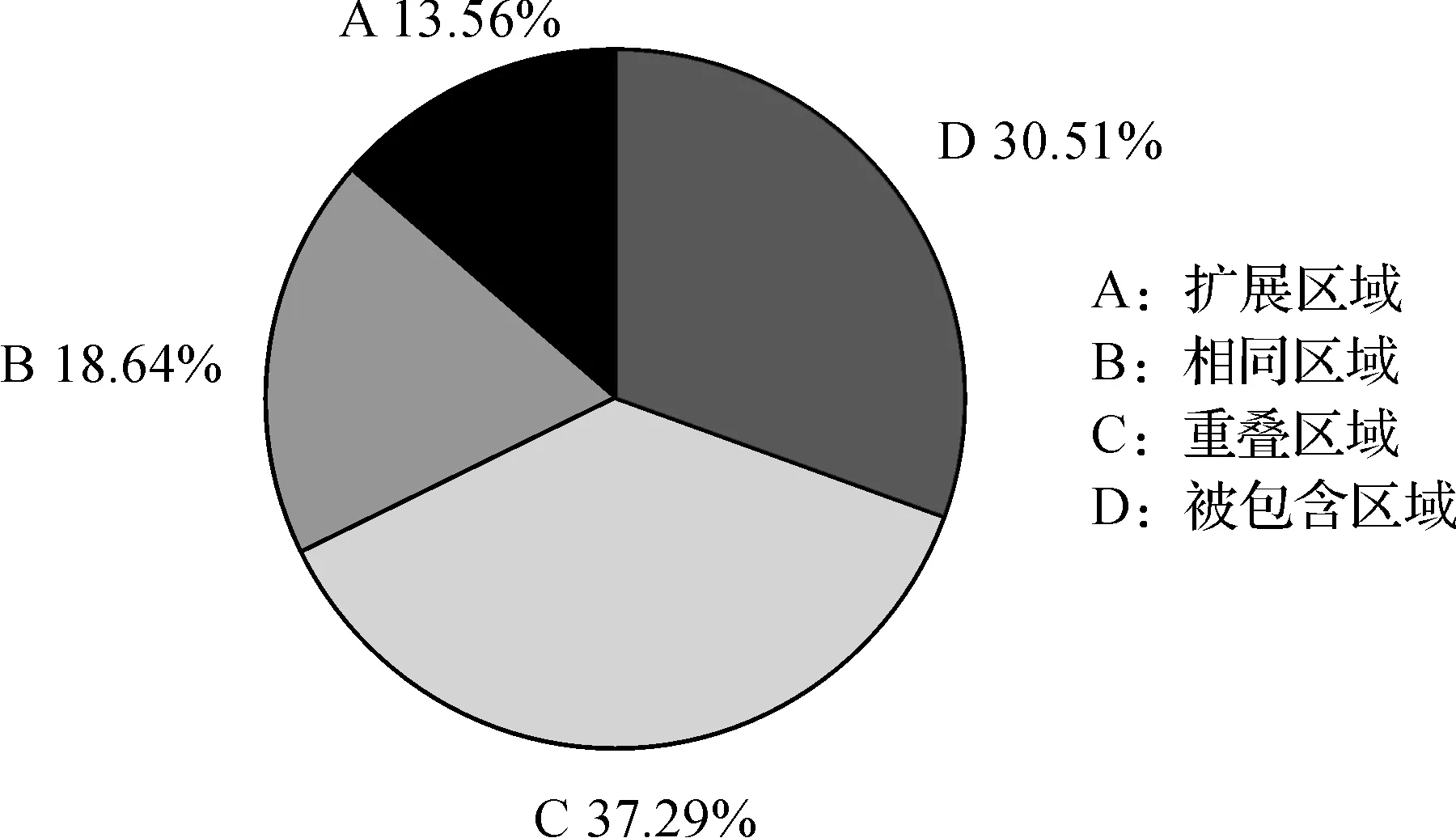
图2 重叠DMRs划分图
为了进一步考查所识别的DMRs的有效性,本文进行如下验证:1)以单个DMR为特征计算原始数据上单个DMR聚类指数,指数越大聚类效果越好.SWKA方法识别的DMRs中有208个都明显大于Ong算法所识别的DMRs聚类指数,如图3(a);2)利用单个DMR为特征对原始数据采用KNN分类,计算LOOCV误差率,误差率越小分类性能越优.SWKA方法发现的DMRs中有208个都明显小于Ong算法识别的DMRS误差率,如图3(b);3)计算SWKA能识别而Ong法未能识别的DMRs聚类指数和分类误差率并与之比较,在两种指标上新识别的DMRs都优于Ong算法识别的DMRs,如图4所示.
不同类别在DMR的维度空间中具有良好的可分性,本文方法识别出的前6个DMRs在不同类别上的平均甲基化水平如图5所示,从图5中可看出,DMR在不同类别上平均甲基化水平存在明显的差异.通过与对照方法的实验结果比较,SWKA方法扩展了对照方法识别出的DMRs长度,比如Ong方法在1号染色体上识别出的一个DMR为[40 090,40 091],而SWKA算法识别的DMR为[40 088,40 089,40 090,40 091],其在4个类别上的平均甲基化水平如图5中的DMR04所示,并且还发现了对照方法未发现的DMRs,比如图5中DMR01,DMR02,DMR03.
综上所述,本文提出的SWKA算法识别的差异甲基化区域在聚类指数和误差率的比较上整体都优于Ong等人提出的DMRs识别算法,也就是说,SWKA算法识别的DMRs比对照算法识别的DMRs差异性更显著.SWKA方法还扩展了对照方法识别出的DMRs长度以及发现对照方法未发现的DMRs,经过真实实验数据上的验证,本文方法准确有效.

图3 SWKA与Ong识别的DMRs的聚类验证和交叉验证结果

图4 SWKA全新识别的DMRs与Ong识别的DMRs的聚类验证和交叉验证结果

图5 DMRs在不同类别上的平均甲基化水平
3结束语
本文在分析最新的DMRs识别算法的基础上,提出了一种利用滑动窗口和KNN分类器识别不同年龄组间存在的DMRs算法.算法能直接运用于多类别的情况,不仅扩展了DMRs长度,还识别出对照的Ong算法未发现的DMRs.本文算法也可以应用于癌症样本的DMRs识别或者其他领域的特征选择问题.
参考文献
[1]杨昆,张彦斌,戴胜冬,等.DNA甲基化的重要特征[J].生物物理学报,2012,28(11):910-922.
[2]JAFFFAE,MURAKAMIP,LEEH,etal.Bumphuntingtoidentifydifferentiallymethylatedregionsinepigeneticepidemiologystudies[J].Internationaljournalofepidemiology, 2012, 41(1): 200-209.
[3]SLIEKERRC,BOSSD,GOEMANJJ,etal.Identificationandsystematicannotationoftissue-specificdifferentiallymethylatedregionsusingtheIllumina450karray[J].Epigenetics&Chromatin, 2013, 6(1): 1-12.
[4]ONGML,HOLBROOKJD.NovelregiondiscoverymethodforInfinium450KDNAmethylationdatarevealschangesassociatedwithaginginmuscleandneuronalpathways[J].AgingCell, 2014, 13(1): 142-155.
[5]ZHANGY,ZHANGJ.Identificationoffunctionallymethylatedregionsbasedondiscriminantanalysisthroughintegratingmethylationandgeneexpressiondata[J].MolecularBioSystems, 2015, 11(7): 1786-1793.
[6]ALISCHRS,BARWICKBG,CHOPRAP,etal.Age-associatedDNAmethylationinpediatricpopulations[J].Genomeresearch, 2012, 22(4): 623-632.
[7]HEYNH,LIN,FERREIRAHJ,etal.DistinctDNAmethylomesofnewbornsandcentenarians[J].ProceedingsoftheNationalAcademyofSciences, 2012, 109(26): 10522-10527.
[8]TROYANSKAYAO,CANTORM,SHERLOCKG,etal.MissingvalueestimationmethodsforDNAmicroarrays[J].Bioinformatics, 2001, 17(6): 520-525.
[9]BOLSHAKOVAN,AZUAJEF.Clustervalidationtechniquesforgenomeexpressiondata[J].Signalprocessing, 2003, 83(4): 825-833.
Algorithm of Identifying Differentially Methylated Region Based on Sliding Windows and KNN
LI Huabing, YANG Kun
(SchoolofComputer,HangzhouDianziUniversity,HangzhouZhejiang310018,China)
Abstract:In view of the shortcomings of the existing methods for identifying differentially methylated regions(DMRs), such as over deletion of sites that significance are weaker, region length limitation and can’t be directly processed by the multi-class. An algorithm of identifying DMRs based on sliding window and k-nearest neighbor(KNN) is proposed. In this method, candidate regions are obtained using sliding windows and KNN, and it merges candidate regions to get DMRs. Through real data simulation results demonstrate the method is superior to control method, such as classification performance, cluster index, the DMRs length of the control methods of Ong is extended and find some DMRs that can’t be found in control algorithm of Ong.
Key words:differentially methylated regions; slide window; k-nearest neighbor classifier; multi-class problem; cluster index
DOI:10.13954/j.cnki.hdu.2016.04.008
收稿日期:2015-11-20
基金项目:国家自然科学基金资助项目(60903086)
作者简介:李华兵(1991-)男,安徽芜湖人,硕士研究生,数据挖掘.通信作者:杨昆副教授,E-mail: yangkun@hdu.edu.cn.
中图分类号:TP391.4
文献标识码:A
文章编号:1001-9146(2016)04-0035-05
