
A2A腺苷受体拮抗剂中间体与抗结剂合成方法研究
2016-07-01 14:41:53屠美玲俞卫平贾继宁张建庭
浙江大学学报(理学版) 2016年4期
屠美玲 ,俞卫平 ,冯 涛 ,贾继宁 ,张 云 ,张建庭
(1.浙江工业大学 化学工程与材料学院, 浙江 杭州 310014; 2. 杭州职业技术学院, 浙江 杭州 310018)
A2A腺苷受体拮抗剂中间体与抗结剂合成方法研究
屠美玲1,俞卫平2,冯 涛2,贾继宁1,张 云1,张建庭1
(1.浙江工业大学 化学工程与材料学院, 浙江 杭州 310014; 2. 杭州职业技术学院, 浙江 杭州 310018)
基于官能化的三唑并[4,5-d]嘧啶类拮抗剂对人体内 A2A腺苷受体拮抗作用的干预治疗,能有效缓解帕金森综合征的临床症状. 该类拮抗剂可以提高多巴胺神经元对纹状体多巴胺的敏感度. 重点研究了三唑并[4,5-d]嘧啶类拮抗剂合成所需的重要中间体4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺的合成、表征及应用. 并对该中间体进行活性拼接,制备了含呋喃基的三唑并[4,5-d]嘧啶类拮抗剂8.
帕金森综合征;A2A腺苷受体拮抗剂;合成
0 引 言
帕金森综合征又名震颤麻痹,是最常见的神经退行性疾病之一. 临床表现包括运动过缓、肌强直、静止性震颤、姿势步态异常等, 也会表现出一定程度的失眠、沮丧以及整体认知能力下降[1-2]. 帕金森综合征由脑黑质中多巴胺神经元的衰退引起,这一衰退导致间质多巴胺浓度降低,从而使基底神经节中神经回路的活力发生了改变[3]. 当帕金森综合征表现出明显的临床症状时,80%的多巴胺神经元已经退化,因此帕金森综合征患病人群多为年纪较大的老人. 在过去60 a中,帕金森综合征的发病率由1/1 000提高到了1/100.
帕金森综合征的临床用药常以左旋多巴(3,4-二羟基-L-苯丙氨酸)制剂为主[4-5].人体大脑中的左旋多巴在多巴脱羧酶作用下发生去羟基化反应释放出多巴胺,从而可以降低神经末梢区域副作用发生的概率.但是这类治疗手段有着重大缺陷:该类药物的药代动力学不稳定,因此难以精确给药剂量.如果被大脑吸收的药物剂量过小就难以释放出足够的多巴胺,易致病情复发;而过大的剂量则会导致无意识的肌肉运动(动作障碍),甚至会出现肌张力障碍、疼痛、身体肌肉局部痉挛等症状.68%的帕金森综合征患者在接受左旋多巴临床治疗后出现了动作障碍后遗症[6].另外,由于反馈抑制作用提高,外界注入左旋多巴会导致内源性多巴胺生成能力的退化.因此通过该疗法很难达到根治目的,并且随着治疗的深入,需要稳步提高给药剂量才能稳固治疗效果.
为了有效弥补这一缺陷,更好地实现对帕金森综合征的治疗,需要寻找一种具有精确药代动力学并且副作用较小的治疗药物. 腺苷是内源性的嘌呤核苷, 存在于所有哺乳类动物组织的细胞外间隙中, 能调节多种生理过程[7]. 在中枢神经系统中, 腺苷可以控制神经递质的释放或突触后神经元的反应, 是一种平衡调节剂. 研究表明,通过人体内A2A腺苷受体的拮抗作用可以减缓帕金森综合征的临床症状[8]. A2A作为腺苷受体家族中的一员(其余3个受体分别为A1、A2B、A3)在中枢神经系统(CNS)中的特定位置表达,并且集中分布于尾状核、壳核、伏核、嗅球以及苍白球的外侧,而这些组织恰恰是多巴胺D2受体集中分布的地方[9]. 大量研究表明,腺苷A2A受体对苍白球神经元的兴奋性作用部分是由于阻止了多巴胺 D2受体的激活. 在制备的纹状体细胞膜上激活腺苷A2A受体会降低多巴胺D2受体的亲和性,这些均说明腺苷A2A受体和多巴胺D2受体有相互拮抗作用. 激活这些腺苷A2A受体会降低D2受体的亲和性并影响细胞内的信号转导通路,从而抑制D2受体的功能. 相反,腺苷A2A受体拮抗剂可以增强多巴胺对这些神经元的功能,从而削弱GABA神经元的抑制性作用,最终增强了丘脑对运动皮层的反馈,实现对帕金森综合征的治疗目的[7, 10].
最早的腺苷A2A受体拮抗剂为KW-6002(该化合物为黄嘌呤衍生物),目前已经完成了帕金森综合征的部分临床试验[11-13]. 但遗憾的是该药在后续关键药效试验测试中表现不佳,没有通过美国FDA认证[11].Vernalis公司后来又开发了2种潜在的腺苷A2A拮抗剂,分别为甲氟喹(2)和三唑并[4,5-d]嘧啶衍生物(3),结构如图1所示. 这2类化合物都有着广泛的应用前景.
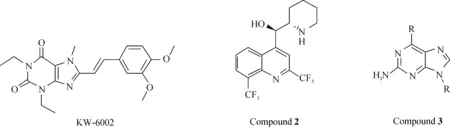
图1 拮抗剂 KW-6002,化合物2,化合物3的化学结构Fig. 1 Structures of antagonist KW-6002, Compound 2 and Compound 3
主要对Vernalis 公司开发的三唑并[4,5-d]嘧啶类腺苷A2A拮抗剂的骨架结构的合成路线进一步优化,采用廉价易得的起始原料,通过4步反应合成关键中间体6-氯-9H-嘌呤-2-胺. 每步反应的收率均超过80%,最终产物纯度达到98%. 在成功获得中间体4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺后,对三唑并[4,5-d]嘧啶类拮抗剂中药效最好的化合物的合成路线进行优化. 大大降低了合成难度,对三唑并[4,5-d]嘧啶类拮抗剂的发展具有一定的推动作用.
1 实 验
1.1 仪器与试剂
NMR光谱用Varian MERCURY plus-400核磁共振(TMS为内标);HPLC测定仪器为岛津LC-2010A;所有溶剂经常规方法纯化(干燥)处理;所有试剂均为市售分析纯或化学纯;柱层析用硅胶为300~400目(青岛海洋化工厂),柱层析用石油醚沸程为60~90 ℃.
1.2 实验方法
1.2.1 4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺(4)的合成
1.2.1.1 2,4-二氨基-6-氯嘧啶(1)
在250 mL 圆底烧瓶中加入75 mL三氯氧磷,并将 2,4-二氨基-6-羟基嘧啶(25 g,198.4 mmol)缓慢加入三氯氧磷溶液中,加入过程中采用磁力持续搅拌反应液. 当2,4-二氨基-6-羟基嘧啶加料结束后,反应溶液快速升温到120 ℃(30 min),回流3 h,减蒸馏除去多余的三氯氧磷,蒸干溶剂后得到含2,4-二氨基-6-氯嘧啶的粗产物. 然后将粗产物倒入200 mL冰水中并用氨水调节溶液pH值在8.5~9.0. 接着用乙酸乙酯(30 mL)萃取分液,收集上层有机相,水相继续用乙酸乙酯萃取(30 mL×3). 合并有机相并用无水硫酸钠干燥、过滤后浓缩得到2,4-二氨基-6-氯嘧啶(26 g,92%收率).1H-NMR (400 MHz, CDCl3) δ∶ 6.57 (2 H, brs), 6.32 (2 H, brs), 5.68 (1 H, s).
1.2.1.2 2,4-二氨基-5-(4-氯苯基)偶氮基-6-氯嘧啶(2)
在250 mL 圆底烧瓶中加入50 mL 稀盐酸溶液(6 mol·L-1),将溶液在冰浴中冷却至0 ℃后缓慢加入对氯苯胺并搅拌10 min. 接着往含有对氯苯胺(12.75 g)的盐酸溶液中滴加亚硝酸钠(6.9 g)水溶液20 mL,滴加完毕后继续搅拌. 反应过程用碘/淀粉试纸监测,当试纸变蓝时意味着反应结束. 反应结束后加入2.5 g尿素以除去体系中未反应的亚硝酸钠,该溶液直接用于下一步反应.
将上述过程制得的重氮盐溶液倒入装有250 mL 2,4-二氨基-6-氯嘧啶(13 g)水溶液的烧杯中,剧烈搅拌30 min后加入醋酸钾(36 g)并继续在室温下搅拌过夜 (18 h). 反应结束后过滤反应液,滤饼用水洗涤并干燥得到2,4-二氨基-5-(4-氯苯基)偶氮基-6-氯嘧啶(22 g,88%产率).1H-NMR (400 MHz, CDCl3) δ: 9.25 (1 H, s), 8.18 (1H, s), 7.81 (2 H, d, J = 8.8 Hz), 7.56 (2 H, d, J = 8.4 Hz), 7.51 (1 H, brs), 7.32 (1 H, brs) ×10-6.
1.2.1.3 2,4,5-三氨基-6-氯嘧啶(3)
将2,4-二氨基-5-(4-氯苯基)偶氮基-6-氯嘧啶(45 g)加入1.5 L甲醇中,然后加入180 mL浓氨水 (25%). 待反应溶液配制完毕后再加入催化剂雷尼镍(18 g)并在氢气(0.101 325 MPa)保护下反应,反应结束后过滤除去催化剂. 滤液减压蒸馏(0.1 MPa)后得到 2,4,5-三氨基-6-氯嘧啶粗产物并用乙醚洗涤5次(50 mL)除去生成的对氯苯胺得到2,4,5-三氨基-6-氯嘧啶的纯品(24 g,94%产率).1H-NMR (400 MHz, CDCl3) δ∶ 6.38 (2 H, brs), 5.52 (2 H, brs), 3.91 (2 H, brs).
1.2.1.4 4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺(4)
在1 L的三口瓶中加入750 mL无水二氧六环,然后加入2,4,5-三氨基-6-氯嘧啶(13.57 g)和亚硝酸异戊酯(12 mL). 待所有物料加入完毕后,将反应瓶中的空气替换为氮气并在90 ℃下反应2 h. 反应结束后将体系冷却到室温并过滤除去固体,滤液用活性炭处理. 接着往活性炭(120目)处理过的二氧六环溶液中加入200 mL正己烷,析出黄色固体(12 g,85%产率).1H-NMR (400 MHz, CDCl3) δ∶ 15.8 (1 H, brs), 7.52 (2 H, brs).
1.2.2 腺苷A2A受体拮抗剂(8)的合成
1.2.2.1 7-呋喃-3H-[1,2,3]三唑并[4,5, d]嘧啶-5-胺(5)
在 250 mL 的三口烧瓶中加入N-甲基吡咯烷酮(97 mL),然后依次加入7-氯-3H-[1,2,3]三唑并[4,5, d]嘧啶-5-胺(13.9 g)、三苯基膦氯化钯(2.8 g)、2-(三丁基锡烷基)呋喃(25.7 mL). 加料完毕后80 ℃反应2 h. 反应过程用TLC监测,当TLC显示反应结束后加入100 mL 乙酸乙酯稀释并过滤后得到含7-呋喃-3H-[1,2,3]三唑并[4,5,d]嘧啶-5-胺的N-甲基吡咯烷酮溶液. 该溶液在202 ℃下浓缩后冷却到室温,加入冰水(200 mL)析出固体,过滤得到7-呋喃-3H-[1,2,3]三唑并[4,5, d]嘧啶-5-胺纯品(15 g,93%产率).1H-NMR (400 MHz, CDCl3) δ∶ 15.57 (1 H, brs),8.11 (1 H, s),7.90 (1 H, d,J=3.6 Hz), 7.23 (2 H, brs), 6.85 (1 H, t, J=1.8 Hz).
1.2.2.2 4-溴甲基-2-甲基-1-硝基苯(6)
在250 mL的三口烧瓶中加入75 mL二氯甲烷,然后将反应瓶置于冰水浴中充分冷却后依次加入3-甲基-4-硝基苯甲醇(9.8 g)、四溴化碳(29.3 g)、三苯基膦(23 g). 加料完毕后用氮气置换烧瓶中的空气并在氮气保护下反应过夜(18 h). 反应结束后减压蒸馏 (0.1 MPa) 除去多余的溶剂,得到4-溴甲基-2-甲基-1-硝基苯粗产物并通过柱层析纯化(乙酸乙酯:石油醚(v∶v=20∶1)得到4-溴甲基-2-甲基-1-硝基苯纯品(12.4 g,92%产率).1H-NMR (400 MHz, CDCl3) δ∶7.97 (1 H, d, J=9.2 Hz), 7.371∶7.372 (2 H, m), 4.46 (2 H, s), 2.61 (3 H, s).
1.2.2.3 中间体(7)
在250 mL的三口烧瓶中加入100 mL DMF,然后依次加入化合物5 (9.55 g)、碳酸钾(6.53 g)后搅拌20 min. 再滴加化合物6 (12 g)的DMF 溶液(20 mL)并继续搅拌2 h. 反应过程通过TLC监测,待TLC显示反应结束后将反应溶液倒入冰水(100 mL)中析出固体,过滤得到中间体8的粗产物并通过柱层析(乙酸乙酯:石油醚(v∶v=20∶1~5∶1)纯化得到纯品(14 g,85%产率).1H-NMR (400 MHz, CDCl3) δ∶8.11 (1 H, d, J=4.4 Hz), 7.96 (1 H, d, J=9.6 Hz), 7.79~7.80 (1 H, m), 7.34 (2 H, s), 6.72~6.74 (1 H, m), 5.70 (2 H, s), 5.44 (2 H, brs), 2.58 (3 H, s).
1.2.2.4 腺苷A2A受体拮抗剂(8)
在1.5 L的三口烧瓶中加入1 L四氢呋喃,然后依次加入前一步合成的中间体7 (8 g)、雷尼镍(1 g). 加料完毕后用氢气置换烧瓶中的空气并在氢气保护下反应过夜(18 h). 反应结束后过滤除去雷尼镍,浓缩滤液得到固体并用四氢呋喃(50 mL×3)洗涤,固体干燥后得到拮抗剂8 (7 g,96%产率).1H-NMR (400 MHz, CDCl3) δ: 8.12 (1 H, s), 7.89 (1 H, d, J = 3.2 Hz), 7.35 (2 H, brs), 6.85~6.93 (3 H, m), 6.55 (1 H, d, J = 8.4 Hz), 5.43 (2 H, s), 4.90 (2 H, s), 2.01 (3 H, s).
2 结果与讨论
对于一种旨在推广的药物来说,成本是制约药物
应用的核心因素之一. 因此在设计该拮抗剂的核心——中间体的反应路线时,笔者选择了廉价易得的起始原料,并对反应过程进行优化,减少一些高成本、高毒性催化剂(例如四三苯基膦钯和钯碳等贵金属)的使用. 在 Vernalis 公司最初的合成路线中采用 2,4-二氨基-6-氯-5-硝基嘧啶作为起始原料,反应得到6-(5-取代)-9H-嘌呤-2-胺,取代后的嘌呤通过 Pd/C 加氢还原硝基得到 2,4,5-三氨基-6-(5-甲基-2-呋喃基)嘧啶,最后发生闭环反应得到 7-取代-1H-[1,2,3]三唑并[4,5,d]嘧啶-5-胺. 具体反应路线如图 2 所示.
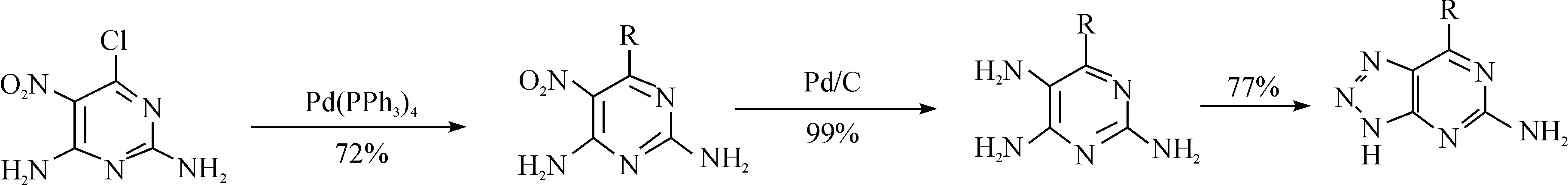
图2 4-取代-1H-[1,2,3]三唑并[4,5,d]嘧啶的传统合成路线Fig. 2 Traditional synthesis strategy of 4-chloro-1H-benzo[d][1,2,3]triazol-6-amine
该路线有2个较大的局限性:(1)起始原料2,4-二氨基-6-氯-5-硝基嘧啶价格昂贵,并且很难获得;(2)在反应中用到了贵金属钯,大大增加了生产成本,同时残留的贵金属也会限制最终拮抗剂作为临床药物的使用. 鉴于此,对该路线进行了一系列优化(见图3).
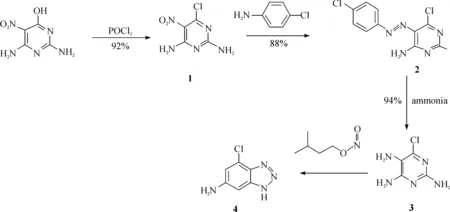
图3 改良4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺合成路线Fig. 3 Improved synthesis strategy of 4-chloro-1H-benzo[d][1,2,3]triazol-6-amine
首先,用对氯苯胺和2,4-二氨基-6-羟基嘧啶为起始原料,设计合理的路线,选择的2种起始原料完全可以代替2,4-二氨基-6-氯-5-硝基嘧啶,大大降低了生产成本.其次,本研究避免使用贵金属催化剂,不但可以降低生产成本,而且能使反应过程更加绿色环保. 由于该中间体含有氯和三唑环上活泼氢2个活性基团,因此通过简单的反应就能合成具有很高生物活性的A2A腺苷受体拮抗剂.
与传统方法相比本合成路线设计更为合理,且具有以下优点:(1)合成原料更加低廉易得.与2,4-二氨基-6-氯-5-硝基嘧啶相比,2,4-二氨基-6-羟基嘧啶不但价格便宜,而且易得. ChemicalBook数据库显示,目前国内市场上有超过 80家的试剂公司可以提供该原料,而2,4-二氨基-6-氯-5-硝基嘧啶在国内则没有供应商. 另一原料对氯苯胺的供应商则超过了100家. (2)该路线摒弃了贵金属催化剂,不但大大节约了生产成本,也克服了体系中的金属残留,为最终药物的推广与应用提供了有利的条件. 与原合成路线相比,本路线更加合理,更具竞争力与环境友好性.
通过上述路线合成得到了重要的中间体4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺. 在 4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺的结构中有3个活性位点,分别是4-位的氯、三唑环上的氮氢、6-位的氨基. 对这3个活性位点的衍生化反应可以制备一系列具有生物活性的腺苷A2A受体拮抗剂. Vernalis公司曾报道过当4-位的氯和三唑环上氢、氮分别被呋喃基与(3-甲基-4-氨基)苄基取代时,该结构具有非常优异的拮抗性,因此笔者在成功制备出中间体4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺的基础上,也研究了该拮抗剂的全合成路线,如图4所示.
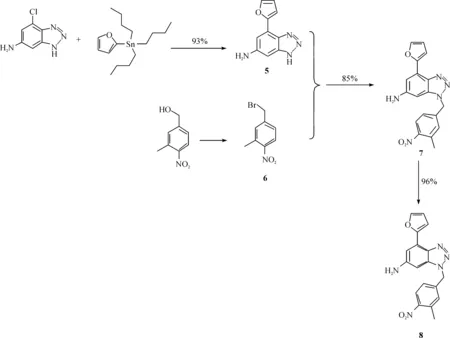
图4 腺苷A2A受体拮抗剂的全合成路线Fig. 4 Synthesis strategy of A2A adenosine receptor
在腺苷A2A受体拮抗剂的全合成中,继续以原料的廉价易得作为第一优化因素. 采用3-甲基-4-硝基苄醇为起始原料制得中间体6. 6继续与中间体5反应得到化合物7,最后通过还原氢化反应得到最终的腺苷A2A受体拮抗剂. 在本合成路线中,每一种原料都廉价易得,且操作简单,每一步反应的收率都超过80%. 值得注意的是,在最后一步还原氢化反应中,仍然没有采用钯碳作为催化剂,其原因有2点:(1)钯碳(Pd/C)价格过高,操作比较复杂;(2)残留的重金属催化剂对人体毒性过大. 因此本文选择雷尼镍作为氢化反应的催化剂,并且获得了较好的结果.
3 结 论
对Vernalis 公司开发的三唑并[4,5-d]嘧啶类腺苷A2A拮抗剂的骨架结构的合成路线进行了优化,采用廉价易得的起始原料,通过4步反应合成关键中间体6-氯-9H-嘌呤-2-胺. 每步反应的收率均超过80%,最终产物纯度达到98%. 在成功获得中间体4-氯-1H-[1,2,3]三唑并[d]嘧啶-6-胺后,还合成了一种拮抗剂8. 本研究优化了合成路线,降低了合成难度,对三唑并[4,5-d]嘧啶类拮抗剂的推广应用具有较大的推动作用.
[1] JANKOVIC J. Parkinson’s disease: Clinical features and diagnosis[J]. J Neurol Neurosurg Psychiatry,2008,79(4):368-376.
[2] BOLLER F , MIZUTANI T , ROESSMANN U,et al. Parkinson disease, dementia, and alzheimer disease: Clinicopathological correlations[J]. Ann Neurol,1980,7(4):329-335.
[3] BLANDINI F, PORTER R H P, GREENAMYRE J T. Glutamate and Parkinson’s disease[J].Mol Neurobiol,1996,12(1):73-94.
[4] COTZIAS G C, PAPAVASILIOU P S, GELLENE R. Modification of Parkinsonism-chronic treatment with L-dopa[J]. New Engl J Med,1969,280(7):337-345.
[5] BARBEAU.L-dopa therapy in Parkinson’s disease: A critical review of nine years’ experience[J]. Can Med Assoc J,1969,101(13):59-68.
[6] RINNE U K. Problems associated with long-term levodopa treatment of Parkinson’s disease[J]. Acta Neurol Scand: Suppl,1983,95:19-26.
[7] 张丹,张建军,刘耕陶.治疗帕金森病新药:腺苷A2A受体拮抗剂[J].中国药理学通报,2006,22(8):897-900.ZHANG Dan, ZHANG Jianjun, LIU Gengtao. A novel therapy for PD-adenosine A2Areceptor antagonist[J]. Chinese Pharmacological Bulletin,2006,22(8):897-900.
[8] SCHIFFMANN S N, HALLEUX P, MENU R, et al. Adenosine A2Areceptor expression in striatal neurons: Implications for basal ganglia pathophysiology[J]. Drug Dev Res,1993,28(3):381-385.
[9] SVENNINGSSONP, LE MOINE C, FISONE G, et al. Distribution, biochemistry and function of striatal adenosine A2Areceptors[J]. Prog Neurobiol,1999,59(4):355-396.
[10] 王恒会,罗蔚锋. 腺苷A2A受体拮抗剂治疗帕金森病的研究进展[J].国外医学:老年医学分册,2006,27(6):244-247.
WANG Henghui, LUO Weifeng. Progress adenosine A2Areceptor antagonist treatment of Parkinson’s disease[J]. Foreign Medical Sciences:Geriatrics,2006,27(6):244-247.
[11] RABASSEDA X, SORBERA L A, MARTIN L , et al. Monographs-kw-6002. Antiparkinsonian, antidepressant, adenosine A2antagonist[J]. Drugs Future,2001,26(1):20-24.
[12] LEWITT P A, GUTTMAN M, TETRUD J W, et al. Adenosine A2Areceptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A doubleblind, randomized, multicenter clinical trial (6002-US-005)[J]. Ann Neurol,2008,63(3):295-302.
[13] HOCKEMEYER J, BURBIEL J C , MUELLER C E. Multigram-scale syntheses, stability, and photoreactions of A2Aadenosine receptor antagonists with 8-styrylxanthine structure: Potential drugs for Parkinson’s disease[J]. J Org Chem,2004,69(10):3308-3318.
Synthesis of an important intermediate of antagonists of the human A2Aadenosine receptor.
TU Meilin1, YU Weiping2, FENG Tao2,JIA Jining1, ZHANG Yun1, ZHANG Jianting1
(1.ChemicalEngineeringandMaterialCollege,ZhejiangUniversityofTechnology,Hangzhou310014,China; 2.HangzhouVocationalandTechnicalCollege,Hangzhou310018,China)
Antagonism of the human A2Areceptor has been implicated as a point of therapeutic intervention in the alleviation of the symptoms associated with Parkinson’s disease. That is to say, at least in part, this kind of antagonists can improve the sensitivity of the dopaminergic neurons to the residual, and deplete levels of striatal dopamine. Herein, we reported a novel synthesis strategy of an important intermediate (4-chloro-1H-benzo[d][1,2,3]triazol-6-amine) of antagonists of the human A2Aadenosine receptor. Additionally, we had also prepared the adenosine receptor 8.
Parkinson’s disease; antagonist of human A2Aadenosine receptor; synthesis

2015-04-07.
屠美玲(1982-),ORCID:http://orcid.org/0000-0002-8700-3524,女,硕士,实验师,主要从事有机中间体研究,E-mail:tu_ml@126.com.
10.3785/j.issn.1008-9497.2016.04.008
TQ 46
A
1008-9497(2016)04-420-06
Journal of Zhejiang University(Science Edition), 2016,43(4):420-425
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
昆明医科大学学报(2021年8期)2021-08-13 08:59:04
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
武警医学(2018年10期)2018-11-06 07:04:34
当代化工研究(2016年6期)2016-03-20 16:21:42
中国果菜(2016年9期)2016-03-01 01:28:41
西华师范大学学报(自然科学版)(2015年3期)2015-02-27 15:31:19
火炸药学报(2014年5期)2014-03-20 13:17:47
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年3期)2014-02-28 17:30:55
