
不同产地太子参芜菁花叶病毒CP基因的分离与序列差异性分析
2016-05-30 09:33:58匡云波叶炜李金辉于静静叶祖云赖钟雄
热带作物学报 2016年10期
关键词:太子参
匡云波 叶炜 李金辉 于静静 叶祖云 赖钟雄

摘 要 通过对不同产地的8份太子参病叶样品进行芜菁花叶病毒(Turnip mosaic virus,TuMV)外壳蛋白(coat protein,CP)基因的RT-PCR检测,从其中7份样品中扩增得到特异性目的条带,对所获得的13个克隆子序列进行下一步的序列分析。结果显示:所得CP基因序列均为775 bp,存在36个核苷酸多态性位点,总变异数为37个,部分点突变仅在某产地发现,可能存在重组;各地分离物之间的遗传分化程度高,遗传漂变可能是造成遗传分化的主要原因;在系统进化树分析结果中,太子参TuMV CP序列均聚类在World B组,并呈现一定的地域特异性。本研究为建立更有效的太子参TuMV检测方法和针对性制定病毒病防控策略奠定重要基础。
关键词 太子参;TuMV;CP;RT-PCR;序列差异性分析
中图分类号 S432.1 文献标识码 A
Abstract RT-PCR method was used to detect 8 infected leaf samples collected from Pseudostellaria heterophylla from different producing areas and 7 samples generated an expected fragment, and 13 clones of the coat protein (CP)genes Tunip mesaic virns(TuMV) were selected for sequence analysis. The results showed that the sequences of all CP genes were 775 bp in length. There existed 36 polymorphic sites, 37 mutations and some of them were geographic region-specific sites. Recombination was found in CP genes. The data showed high genetic differentiation among the isolates from Guizhou and Shandong, Jiangsu and Shandong provinces. Genetic drift could be the main cause of genetic differentiation. All the CP genes obtained in this study were classified into World B group in the phylogenetic tree, and indicated somehow geographic region specificity. The results reported in this manuscript laid an important foundation for the establishment of a more efficient molecular detection method and the targeted strategy for the control of TuMV disease in P. heterophylla.
Key words Pseudostellaria heterophylla;TuMV;CP;RT-PCR;Sequence difference analysis
doi 10.3969/j.issn.1000-2561.2016.10.021
太子参(Pseudostellaria heterophylla),又名孩儿参、童参,为石竹科太子参属多年生草本药用植物,收载于中华人民共和国药典中,具有益气健脾、生津润肺之功效。太子参在中国华东、华中、华北和西北等地都有分布,主产于福建、贵州、江苏、山东、湖南等地,栽培历史已过百年。长期以来,太子参均以块根进行无性繁殖,导致病毒不断积累和扩散,太子参种性退化,病毒病害日趋严重。据报道,已知侵染太子参的病毒病病原有4种,其中芜菁花叶病毒(Turnip mosaic virus,TuMV)是侵染太子参的主要病毒病病原,分布于江苏和山东的各个太子参产区[1]。
TuMV 属于马铃薯 Y 病毒属(Potyvirus)成员,可通过汁液或蚜虫传播,在自然条件下主要靠蚜虫以非持久性方式传播。TuMV是RNA病毒,其寄主范围广泛,而且大部分为十字花科作物。近年来,关于TuMV的分子进化和遗传结构的研究已经取得了较大的研究进展[2]。大量研究结果表明,由于地理起源和株系特化等原因,TuMV易产生变异,导致TuMV存在多种株系和致病型[3-4]。TuMV分离物划分为2个致病型:B型和BR型。B型只侵染芸苔属(Brassica),BR型既侵染Brassica,又侵染萝卜属(Raphanus)[3]。根据外壳蛋白(coat proein,CP)等基因进行系统发育分析,TuMV分离物大致分为Basal-B、Basal-BR、Asian-BR和World-B组[3-4]。
迄今为止,有关太子参TuMV的鉴定与检测仅以病毒形态学、生物学和血清学特征为主要依据[1,5-7]。随着分子生物学的发展,植物病毒的鉴定所需要的信息更全面、更精确,有必要对太子参TuMV进行进一步的分子鉴定。鉴于此本研究拟对来自于不同产地的太子参病毒病病样进行TuMV CP基因的RT-PCR扩增和克隆,以了解TuMV在太子参产地的发生、分布情况;同时对所得病毒序列进行遗传多样性、分子进化等相关的生物信息学分析,以探讨地理因素对太子参TuMV遗传特性的影响,为太子参TuMV病毒的分子检测、病毒病防控提供有效的理论依据。
1 材料与方法
1.1 材料
供试毒源为2015年3月取自于福建、江苏、湖南、贵州、山东的太子参病叶,观察到植株矮小、叶片皱缩、伴有重花叶等典型症状,经液氮速冻后于-80 ℃保存备用。
1.2 方法
1.2.1 病叶总RNA的提取 采用Trizol试剂(Invitrogen)按照说明书提取太子参病叶总RNA。采用1%琼脂糖凝胶电泳检测RNA的质量,并采用核酸测定仪测定RNA的浓度。
1.2.2 引物设计 根据GenBank数据库中的TuMV CP的核苷酸序列的保守区域,采用Primer 5.0软件设计1对简并引物:上游引物TuMV-F为5′-AGGTGAAAYGCTTGATGCAGGTY-3′,下游引物TuMV-R为5′-GTTHCCATCCARKCCGAACAAAT-3′。引物由北京六合华大基因科技股份有限公司进行合成。
1.2.3 CP基因的克隆 取病叶总RNA 5 μg,采用RevertAid First Strand cDNA Synthesis Kit(Fermentas)进行cDNA第一链的合成(10 μL体系)。以反转录产物为模板,进行PCR扩增。PCR 反应体系为:cDNA 模板1 μL,上、下游引物各1 μL(10 μmol/L),10×Taq Plus Buffer 2.5 μL,dNTP Mix(2.5 mmol/L)4 μL,Taq Plus(2.5 U/μL)0.25 μL,加H2O至总体积25 μL。PCR扩增条件为:94 ℃预变性4 min;94 ℃变性30 s,53 ℃退火30 s,72 ℃延伸50 s,共35个循环;最后72 ℃延伸10 min。PCR扩增所采用的试剂为2×Taq Plus PCR Master Mix(TianGen)。取3 μL PCR扩增产物进行琼脂糖(1%)电泳检测。采用DNA快速纯化回收试剂盒(Solarbio)回收目的片段,以pMD-18T(TaKaRa)连接载体,Escherichia coli(DH5α)感受态(TianGen)对回收片段进行连接、克隆和转化,阳性克隆子送北京六合华大基因科技股份有限公司进行测序。
1.2.4 序列分析 测序获得的核苷酸序列及其推导的氨基酸序列在NCBI的GenBank数据库中进行在线Blast,相关序列登录GenBank。采用DNAMAN软件进行序列比对(不含引物序列),利用DnaSP v.5.10.1软件进行DNA多样性分析、同义与非同义替换估算、基因重组、遗传分化与基因流分析等,统计核苷酸多样性(π)、核酸单体型多态性(Hd)、遗传分化系数(Fst)和基因流(Nm)等参数。使用MEGA 6.06软件中的Neighbor-Joining(邻位相连法,NJ)法建立系统发育树(P-distance法),并应用bootstrap法(重复1 000次)评估系统发育树。
2 结果与分析
2.1 不同产地太子参TuMV CP基因的克隆
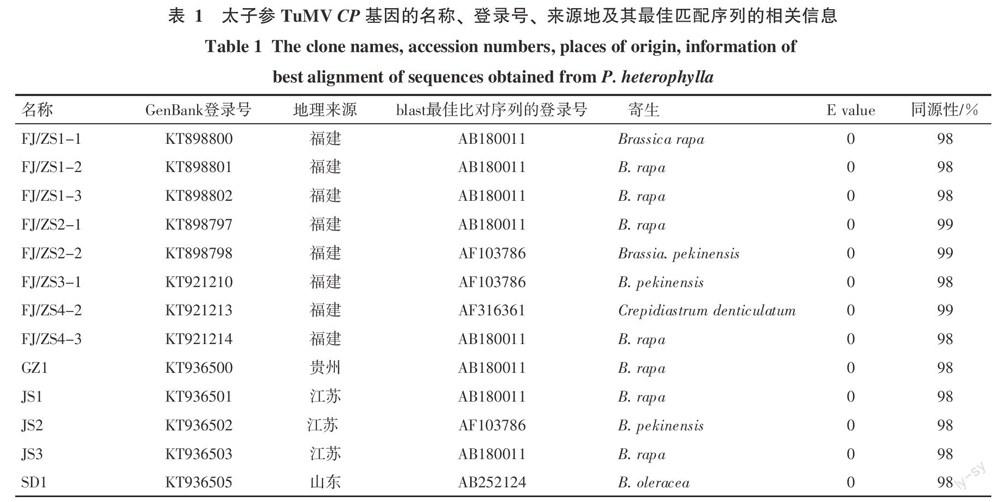
以健康太子参为对照,采用TuMV病毒CP基因的间并引物对8份不同产地的太子参病样进行PCR扩增,其中除样品“HN”无扩增条带外,其他7份样品均获得了1条明亮、清晰的大小约为775 bp的特异性条带,符合预期目的条带大小(图1)。对目的条带进行回收纯化、克隆和测序,来自每个样品的测序克隆数为3~5个。将测序得到的cDNA片段的核苷酸序列及其推导的氨基酸序列在NCBI的GenBank数据库中在线进行Blast,结果显示与其它植物TuMV CP基因在核苷酸序列和氨基酸序列上均具有很高的同一性,这表明所得片段为太子参TuMV CP基因的部分序列。
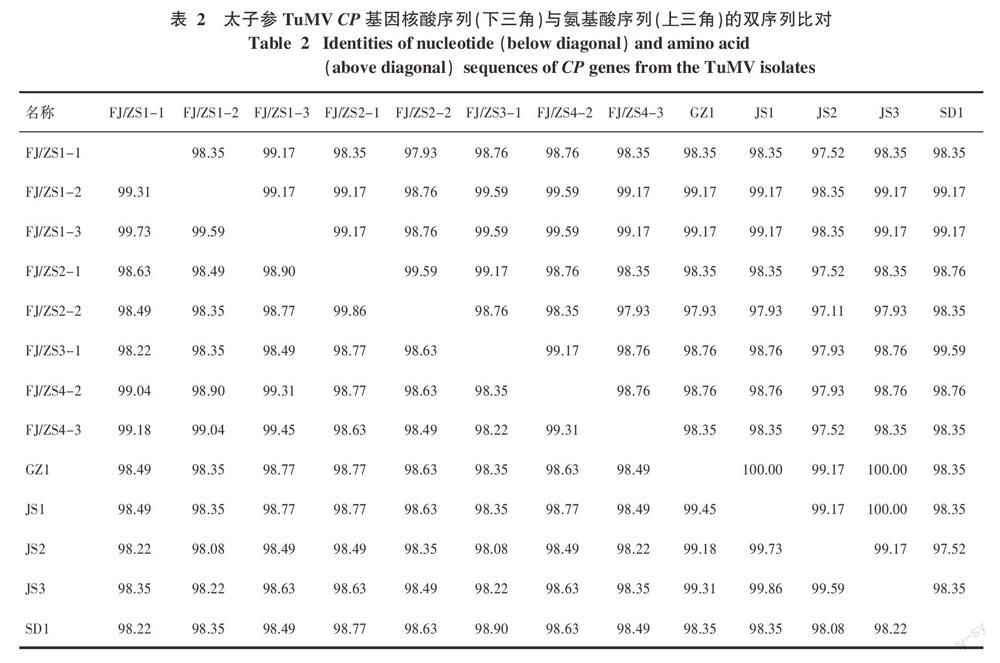
结果表明,7个太子参病样的CP扩增产物共获得20个克隆序列,均为775 bp。去掉引物序列后,筛选其中具有序列差异的13个克隆,进行下一步的序列分析,其克隆序列的名称、GenBank登录号、地理来源、blast最佳比对序列的登录号、E value和同源性见表1。太子参TuMV CP序列与NCBI上登录的黄瓜假还阳参(Crepidiastrum denticulatum,AF316361)分离物等同源性最高,达99%。双序列比对结果显示,来自不同样品的克隆间核苷酸和氨基酸序列的同源性分别在98.08%~99.45%和97.52%~100%;来自同一样品扩增产物的克隆序列同源性更高,其核苷酸和氨基酸序列同源性分别在99.31%~99.86%和98.35%~100%(表2)。
2.2 不同产地太子参TuMV CP基因的遗传多样性分析
将上述太子参TuMV CP基因核苷酸序列导入DnaSP v.5.10.1中。经DNA多样性分析,结果发现其总单倍型多样性Hd为1.000,总多态性指数Pi为0.013 14;无序列插入或缺失现象。存在36个多态性位点,其中单一多态位点20个,简约信息位点16个;总变异个数为37,其中10个同义突变,27个异义突变;部分点突变仅在某区域性产地发现,如江苏分离物(第64、701个位点)、山东分离物(第165、368、677个位点)、贵州分离物(第419个位点)等。进行同义与非同义替换估算,结果显示,同义位点数为157.32,非同义位点数为571.68,同义突变多样性πsyn=0.012 55小于非同义突变多样性πnonsyn=0.013 30,说明CP基因同义突变的遗传变异程度小于非同义突变。经重组分析结果发现,在太子参分离物CP基因上发生的最小遗传重组事件数为4个,分别在以下位点之间:92~125、125~203、263~341、341~686。
将太子参TuMV CP基因序列按地理来源分为福建分离物、贵州分离物、江苏分离物、山东分离物4组以进行遗传分化和基因流分析。福建分离物组内病毒CP基因的核苷酸多样性和单倍体多样性均为最高。不同分离物间总的遗传分化系数Fst为0.750 93,总基因流Nm为0.17,暗示各地分离物间遗传分化程度高,基因交流不频繁,容易发生遗传漂变。各地分离物的遗传分化系数Fst在0.544 74~1.000 00,其中贵州分离物和山东分离物之间的Fst值最高,为1.000 00,说明这2个地区间遗传分化程度相对较高。
2.3 TUMV CP基因的分子进化分析
应用MEGA 6.06软件,对所得13条太子参TuMV CP基因序列与已报道的其他寄主植物TuMV CP序列进行系统进化分析,结果见图2,TuMV病毒分离物可以分为Basal-BR、World-B、Asian-BR、Basal-B 4个组,每个组内均包含了不同寄主植物的TuMV序列,与Ohshima等[3]的研究结果基本一致。太子参TuMV序列与PV0054、KEN1、USA1、GBR7聚类在World-B组,可能属于B致病类型。不同产地太子参TuMV CP基因序列进一步分成3个小分支,呈现明显的地域特异性。贵州的GZ1与江苏的JS1、JS2、JS3聚在一个分支。福建的FJ/ZS1和福建的FJ/ZS4聚在一个分支,而山东的SD1与福建的FJ/ZS2、FJ/ZS3分离物聚在一个分支,与FJ/ZS3亲缘关系最近。来自福建的4个分离物之间也存在着一定的遗传差异,可能是因为来源于不同太子参品种的原因。
3 讨论与结论
本研究采用简并引物对不同产地的8个太子参病样进行RT-PCR检测,除来自湖南的样品“HN”外,其他7个样品均扩增到了特异目的条带,说明TuMV分布范围很广,福建、贵州、江苏、山东均存在TuMV侵染太子参的情况,而湖南可能存在其他花叶病病毒侵染。
中国地域辽阔,地形复杂,生境多样,存在多种TuMV变异类型[8-10]。首先,从系统进化树分析看,所有TuMV CP序列分为Basal-BR、World-B、Asian-BR、Basal-B 这4个组,每个组内均包含了不同寄主植物的TuMV序列,无明显的寄主特异性。本研究获得的不同地理来源的太子参TuMV CP序列均属于World-B组。据报道,World-B组包含的TuMV病毒大多为B致病型,少数为BR致病型[3]。但宋云枝[8]从山东省3个地市感病的白菜和萝卜上分离的6个TuMV分离物属于World-B组,均被鉴定为BR致病型。太子参TuMV的致病类型还有待进一步研究确定。其次,太子参TuMV CP基因序列的聚类具有明显的地域特异性。虽然不同产地太子参TuMV CP基因序列的同源性达到了98%以上,具有高度的保守性,但仍具有一定的分子变异。本研究结果发现不同产地太子参TuMV CP基因序列存在36个多态性位点,37个突变,其中10个同义突变,27个异义突变。这些突变体都是通过核苷酸碱基转换或颠换形成的,未出现序列插入或缺失现象,而且有些突变是某产地所特有的,如江苏分离物、山东分离物、贵州分离物均发现1~3个地区特异性点突变,说明不同产地太子参TuMV间存在一定的地理间隔。
不同地理来源的太子参TuMV分离物CP基因间可能存在遗传重组。据报道,遗传重组是马铃薯Y病毒属病毒变异的一个重要因素,TuMV含有多个重组热点,多数重组发生在P1基因和CI/6K2/VPg基因区域[11],只有极少数重组发生在CP和3′ UTR区域[10,12]。无论在植物还是动物RNA病毒中,重组都是很常见的现象,被认为是病毒毒性改变的决定因素,涉及新的病毒株系的出现[11,13-14]。贵州分离物与山东分离物、江苏分离物与山东分离物之间遗传分化程度最高,遗传漂变可能是造成遗传分化的主要原因。
本研究通过RT-PCR扩增获得了不同产地太子参TuMV的CP基因序列,并进一步进行了相关的序列分析,探明了太子参各产地TuMV的发生、分布情况,阐明了太子参TuMV CP基因的分子变异、遗传分化程度与归属类群,为建立太子参TuMV的特异性分子检测方法和针对性制定病毒病防控策略奠定了重要基础。
参考文献
[1] 宋荣浩, 濮祖芹. 太子参(Pseudostellaria heterophylla)病毒病病原鉴定[J]. 上海农业学报, 1991, 7(2): 80-85.
[2] Ohshima K. Plant potyvirus evolution: the survey of the genetic structure of populations[J]. Uirusu, 2012, 62(2): 151-160.
[3] Ohshima K, Yamaguchi Y, Hirota R, et al. Molecular evolution of Turnip mosaic virus: evidence of host adaptation, genetic recombination and geographical spread[J]. J Gen Virol, 2002, 83(6): 1 511-1 521.
[4] Yasaka R, Ohba K, Schwinghamer M W, et al. Phylodynamic evidence of the migration of Turnip mosaic potyvirus from Europe to Australia and New Zealand[J]. J Gen Virol, 2015, 96(3): 701-713.
[5] 陆家云. 药用植物病害[M]. 北京: 中国农业出版社, 1995.
[6] 黄勇毅,林丛发. 闽东太子参花叶病发病规律调查及防治途径研究[J]. 宁德师专学报(自然科学版), 2004, 16(1): 65-68.
[7] 刘清琪,陈棣华. 太子参花叶病病原及其防治的初步研究[J]. 中药材科技, 1983, 6(2): 11-12.
[8] 宋云枝. 芜菁花叶病毒山东分离物外壳蛋白基因的克隆及序列分析[J]. 中国农业科学, 2005, 38(3): 504-510.
[9] 张成玲. 不同寄主植物上芜菁花叶病毒外壳蛋白基因克隆及分析[J]. 江苏农业学报, 2014, 30(1): 37-41.
[10] 田延平. 芜菁花叶病毒不同分离物3′-cDNA片段的克隆及序列分析[J]. 植物病理学报, 2005, 51(S1): 38-42.
[11] Tan Z Y, Wada Y, Chen J S, et al. Inter- and intralineage recombinants are common in natural populations of Turnip mosaic virus[J]. J Gen Virol, 2004, 85(9): 2 683-2 696.
[12] Tan Z Y, Gibbs A J, Tomitaka Y, et al. Mutations in Turnip mosaic virus genomes that have adapted to Raphanus sativus[J]. J Gen Virol, 2005, 86(Pt 2): 501-510.
[13] Ohshima K, Tomitaka Y, Wood J T, et al. Patterns of recombination in Turnip mosaic virus genomic sequences indicate hotspots of recombination[J]. J Gen Virol, 2007, 88(1): 298-315.
[14] Han Z X, Zhang T T, Xu Q Q, et al. Altered pathogenicity of a tl/CH/LDT3/03 genotype infectious bronchitis coronavirus due to natural recombination in the 5′-17kb region of the genome[J]. Virus Res, 2015, 213(1): 140-148.
猜你喜欢
现代农业科技(2022年3期)2022-02-21 06:21:30
花卉(2021年14期)2021-07-26 02:22:38
消费导刊(2016年3期)2016-04-13 03:15:21
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
中国现代中药(2011年4期)2011-09-27 11:40:49
中国现代中药(2011年8期)2011-02-10 07:21:51
