
含能材料中间体3,7,10-三氧代-2,4,6,8,9,11-六苄基-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HBPTO)的合成、表征及工艺改进
2016-05-09 02:48王锡杰毕福强王伯周张俊林
含能材料 2016年8期
王锡杰, 毕福强, 肖 川, 王伯周, 张俊林, 周 诚, 胡 银
(西安近代化学研究所, 陕西 西安 710065)
1 引 言
螺桨烷是一种特殊的稠合三环体系,三环共用一个C—C共价键,其空间张力很大,反应活性很强,具有重要的生物和药理活性,是构建许多天然产物的基本骨架结构[1-3],在过去的30年里,因其独特的结构吸引了诸多化学工作者的兴趣[4-8]。而氮杂[3.3.3]螺桨烷由于具有较多的氮原子数、较好的对称性、紧凑的骨架,使其密度较高,同时其空间张力大有助于提高生成焓、爆速及作功能力,结构中的氮原子也为进一步引入含能基团提供了活性反应位点,适宜作为含能化合物的骨架结构。近年,含能材料科研工作者已将氮杂[3.3.3]螺桨烷结构应用于含能材料的设计与合成中[9],理论研究结果表明[9-12],硝基氮杂[3.3.3]螺桨烷可能突破传统CHON材料的能量阈值,同时保持一定的稳定性和安全性。3,7,10-三氧代-2,4,6,8,9,11-六苄基-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HBPTO)是韩国国防局于2013年最新报道一种螺桨烷化合物[13],其合成以尿酸(UC)为起始原材料,经氧化-取代反应合成二氨甘脲(DAGU),DAGU再经缩合、原位脱BOC保护二步反应合成3,7,10-三氧代-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(PTO),再经取代反应共计四步合成目标化合物HBPTO。在HBPTO的环上引入叠氮基、硝基及氨基等基团设计合成新型高能量密度材料,如2,4,6,8,9,11-六硝基-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HNHAP),其理论密度为2.01 g·cm-3,理论爆速9665 m·s-1,理论爆压45.0 GPa,氧平衡为-3.75%,综合性能优异,在武器装备中具有潜在的应用前景。
本研究参考文献[13],以尿酸为起始原材料,经氧化-加成、缩合、取代反应等在国内首次合成了HBPTO(Scheme 1),并采用红外光谱、核磁共振、质谱以及元素分析对中间体和目标化合物的结构进行了表征。针对PTO二步合成方法存在工艺路线较长、工艺操作复杂等问题,对PTO的合成工艺进行了重大改进,自行设计了一步缩合反应合成PTO的新方法,缩短了反应步骤、简化了操作工艺,且收率较高。首次探讨了DAGU合成反应机理,优化了合成工艺条件,确定了最佳合成工艺参数。
2 实验部分
2.1 仪器与试剂
德国Bruker公司TENSOR 27型傅里叶变换红外光谱仪; 德国Elementar公司Vario EL Ⅲ型自动微量有机元素分析仪; 瑞士Bruker公司AV 500型(500 MHz)超导核磁共振波谱仪; 美国Waters公司的SYNAPT型UPLC-Q-TOFMS液质联用仪; 日本岛津GC-2010型高效液相色谱仪。
尿酸(≥98%),分析纯,东京化成工业株式会社; 铁氰化钾(≥99.5%),分析纯,郑州派尼化学试剂厂; 氨水(25%-28%),分析纯,西陇化工股份有限公司; 羰基二咪唑(≥97%),分析纯,Aladdin Industrial Coporation; 苄溴,化学纯; 二甲亚砜(DMSO)、二甲基甲酰胺(DMF) 、氢化钠(≥60%)均为分析纯,均为成都市新都区木兰镇工业开发区生产厂。
2.2 实验
2.2.1合成路线

Scheme 1 Synthetic route of HBPTO
2.2.23a,6a-二氨基四氢咪唑并[4,5-d]咪唑-2,5(1H,3H)-二酮(DAGU)的合成
室温下,将40.36 g(23.4 mmol)尿酸与153.6 mL氨水、408 mL水混合后,冷却至0 ℃后,温度保持在0~5 ℃分批加入316.8 g(96.3 mmol)铁氰化钾,加料完毕后,升温至25 ℃反应0.5 h,过滤,滤液放置2天后析出白色固体; 滤饼用冰水洗涤至水为无色,然后将滤饼加入到50 mL的氨水中,常温下搅拌反应12 h,过滤,水洗,乙醇、乙醚洗,与滤液析出的白色固体合并干燥,得DAGU固体17.9 g,收率43.2%。1H NMR (DMSO-d6, 500 MHz),δ: 2.35(S,4H), 7.04(S,4H);13C NMR (DMSO-d6, 125 MHz),δ: 157.88, 87.28; IR (KBr,ν/ cm-1), 3349, 3302, 1737, 1687, 1489, 1208, 927; MS(ESI)m/z(%): 173(M+H)+; Anal.calcd for C4H8N6O2: C 27.91, H 4.65, N 48.84 ; found C 27.89, H 4.67, N 48.38。
2.2.33,7,10-三氧代-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷的合成(PTO)
氮气保护下,将二氨基甘脲6 g(35.0 mmol)与羰基二咪唑6 g(37.0 mmol)分别加入到123 mL DMSO中,室温下反应60 h,反应完毕后,将反应液倒入到530 mL丙酮中,析出白色固体,过滤,甲醇洗涤,干燥得5.96 g PTO产品,收率85.6%。1H NMR (DMSO-d6, 500 MHz),δ: 8.04(s);13C NMR (DMSO-d6, 125 MHz),δ: 159.57, 85.25; MS(ACPI)m/z(%): 197(M-H)-。
2.2.43,7,10-三氧代-2,4,6,8,9,11-六苄基-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HBPTO)的合成
氮气保护下,将PTO 0.45 g(2.27 mmol)加入到5 mL DMF和2 mL DMSO中,冷却至0 ℃后,加入NaH 0.85 g(21.27 mmol)后,0 ℃反应1 h,再加入卞溴3.4 mL(28.28 mmol),升至常温,反应12 h后,倒入水中,乙酸乙酯萃取,蒸馏,柱分离后得0.32 g HBPTO产品,收率23.2%,纯度98.5%(HPLC)。1H NMR (DMSO-d6, 500 MHz),δ: 7.29-7.23(m, 18H), 7.11-7.09(m, 12H), 4.32(s, 12H);13C NMR (DMSO-d6, 125 MHz),δ: 157.1, 136.7, 128.6, 127.6, 126.4, 89.3, 45.6; IR (KBr,ν/cm-1):3088, 3062, 2967, 2929, 1718, 1471, 1452, 1425, 1333, 1142, 1075; MS(ACPI)m/z(%): 739.3394(M+H)+; Anal.calcd for C47H42N6O3: C 76.40, H 5.73, N 11.37; found C 76.01, H 5.63, N 10.92。
3 结果与讨论
3.1 谱学解析
3.1.1 红外光谱解析
HBPTO的红外光谱见图1。图1中,3088 cm-1,3063 cm-1为苯环上CH键的伸缩振动,1355 cm-1为苯环上CH键的弯曲振动, 2929 cm-1为苄基CH2键伸缩振动,1452 cm-1苄基CH2的弯曲振动,1718 cm-1为羰基的伸缩振动。1496 cm-1,1471 cm-1为苯环骨架振动, 738,699 cm-1为苯环上CH面外弯曲振动,红外光谱反映了HBPTO结构中的苯环、亚甲基、次甲基特征官能团的存在。
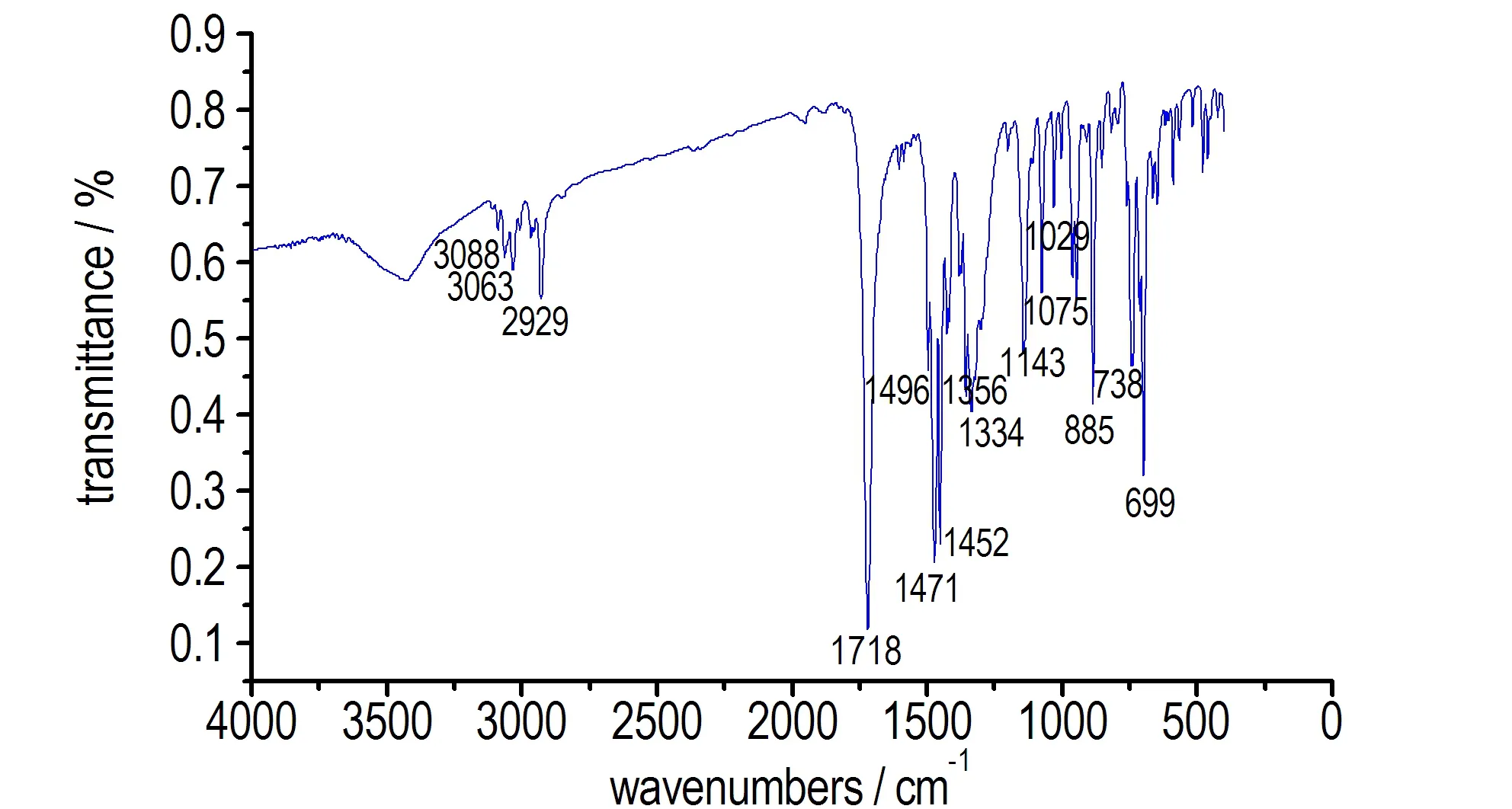
图1HBPTO的FT-IR图
Fig.1FT-IR spectrum of HBPTO
3.1.2 质谱解析
HBPTO的大气化学电离质谱(APCI-MS) 如图2所示。采用液相色谱串联质谱,选择正离子模式测定,并通过已知内标物校正,获得HBPTO的精确分子量,谱图中m/z739.3394恰好与HBPTO分子量相差1,判断m/z739.3394为样品的准分子离子峰(M+H)+,m/z761.3216恰好与HBPTO分子量相差23,判断m/z761.3216为样品的(M+Na)+峰,碎片m/z563.2896正好为m/z739.3394失去一个H,三个CO,一个Bn形成的碎片离子,碎片m/z290.2875正好为碎片m/z563.2896失去三个Bn形成的碎片离子,而碎片m/z91.0714为Bn碎片离子。

图2HBPTO的质谱图
Fig.2Mass spectrum of HBPTO
3.1.3 核磁共振光谱
3.1.3.11H NMR谱
HBPTO的质子结构及化学位移归属如图3 (1H NMR谱图)所示。由图3可见, 3组氢共42个质子,其积分比(由高场到低)约为18∶12∶12。
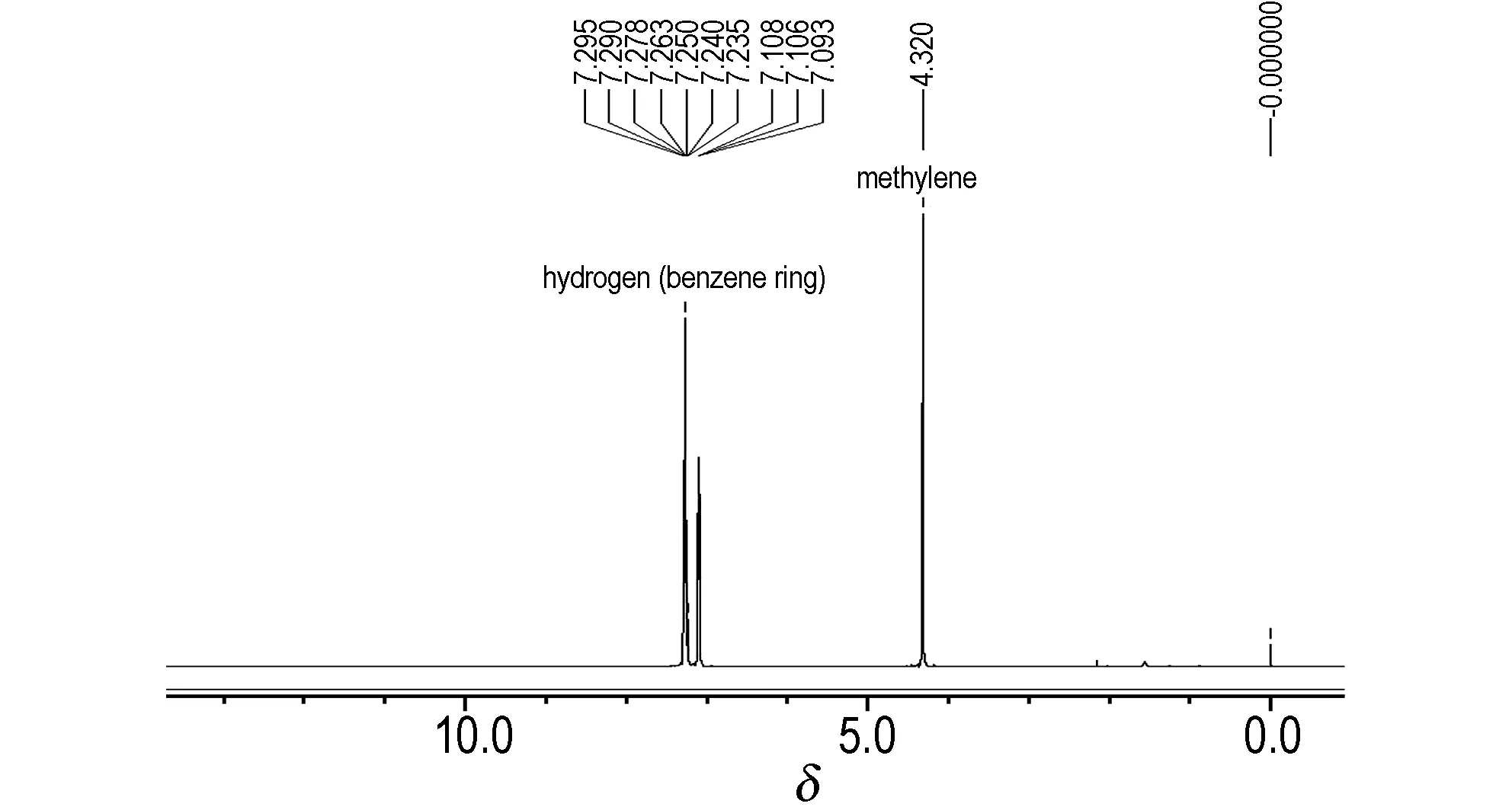
图3HBPTO的1H NMR谱图
Fig.31HNMR spectrum of HBPTO
3.1.3.213C NMR谱
HBPTO的13C NMR谱图如图4所示。图4显示,HBPTO为对称结构,共有7个吸收峰,表明分子中有7种处于不同化学环境下的碳原子。δc157.281为螺桨烷上的—CO 碳,δc136.831为苯环上的季碳,δc128.776为苯环上的间位碳,δc127.741为苯环上邻位碳,δc126.572为苯环上的对位碳,δc89.472为螺桨烷上的季碳,δc45.769为苄基上的—CH2由以上分析表明,其碳原子种类数及化学位移值与预定分子一致。
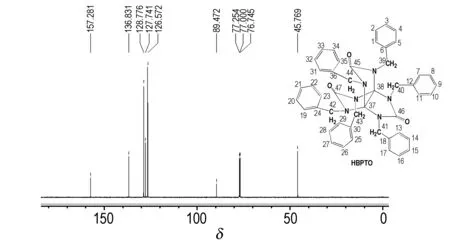
图4HBPTO的13CNMR谱图
Fig.413CNMR spectrum of HBPTO
3.2 DAGU反应机理
尿酸环内双键结构在氨水体系中首先与一分子氨发生双键加成反应,得到顺式5,6-双环结构Ⅰ,其中酰胺羰基较之脲羰基活性较高,碱性环境下,氢氧根负离子进攻酰胺羰基结构之后水解开环得到羧酸负离子,在氧化剂存在条件下羰基负离子被氧化得到自由基,继而诱导脱羧形成不饱和烯胺化合物Ⅲ,其同分异构体Ⅳ与氨发生1,4-加成反应得到化合物Ⅴ,Ⅴ分子内脲胺基发生5-exo的1,2-加成得到DAGU。推测其反应机理如Scheme 2所示。

Scheme 2 Possible mechanism of synthesizing DAGU by uric acid
3.3 DAGU工艺优化
3.3.1 氧化剂使用量的影响
反应温度为25 ℃,反应时间为0.5 h的条件下,考察氧化剂K3Fe(CN)3加入量(n(UC)∶n(K3Fe(CN)3)分别为1∶2, 1∶3, 1∶3.5, 1∶4, 1∶4.2, 1∶5, 1∶6)对DAGU收率及纯度的影响,实验结果见表1。从表1可见,n(UC)∶n(K3Fe(CN)3)为1∶4.2时,收率、纯度最高,这是由于理论上铁氰化钾的摩尔量是DAGU的两倍,且该反应为非均相反应,在反应温度不高的情况下,反应活性差,因此铁氰化钾的用量较理论量高,n(UC)∶n(K3Fe(CN)3)=1∶4.2为较佳用量,进一步提高铁氰化钾使用量,收率未见提高。
表1n(UC)∶n(K3Fe(CN)3)对DAGU收率的影响
Table1Effectofn(UC)∶n(K3Fe(CN)3)ontheyieldofHBPTO
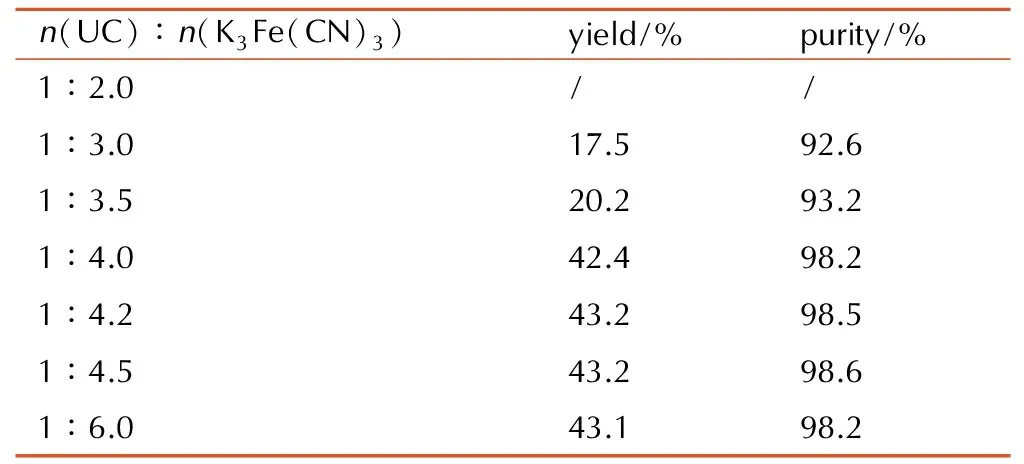
n(UC)∶n(K3Fe(CN)3)yield/%purity/%1∶2.0//1∶3.017.592.61∶3.520.293.21∶4.042.498.21∶4.243.298.51∶4.543.298.61∶6.043.198.2
3.3.2 反应温度的影响
n(UC)∶n(K3Fe(CN)3)∶1∶4.2,反应时间为0.5 h的条件下,考察温度对DAGU收率的影响,结果见表2。从表2 可见,当反应温度为25 ℃时,反应收率最高,温度进一步提高时,反应收率降低,这是因为当温度提高时,产物DAGU易水解,造成收率与纯度的下降。故25 ℃为适宜的反应温度。
表2反应温度对DAGU收率的影响
Table2EffectofreactiontemperatureontheyieldofDAGU
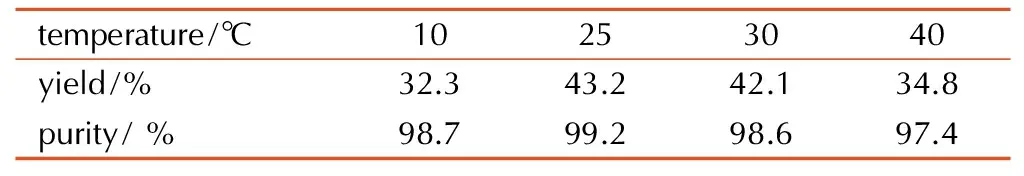
temperature/℃10253040yield/%32.343.242.134.8purity/%98.799.298.697.4
3.3.3 反应时间的影响
n(UC)∶n(K3Fe(CN)3)为1∶4.2,反应温度为25 ℃的条件下,考察反应时间对DAGU收率的影响,结果见表3。从表3可见,当反应时间为0.5 h,反应收率最高,反应时间进一步延长,收率没有明显提高,当反应时间为3 h,收率反而有所降低,这是因为反应时间过长,产物DAGU溶解于水中可能会产生一定水解所致。因此,最优反应时间为0.5 h。
表3反应时间对DAGU收率的影响
Table3EffectofreactiontimeontheyieldofDAGU

time/h0.513yield/%43.243.142.1purity/%99.299.299.0
3.4 PTO工艺优化
3.4.1 PTO工艺改进
文献[13]在2013年公开了3,7,10-三氧代-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷的合成方法,其合成路线见Scheme 3。

Scheme 3 Two-step synthetic route of PTO
该方法分为两步,第一步DAGU与二碳酸二叔丁酯发生缩合反应后,采用乙醚萃取,蒸馏后,色谱柱纯化后,得到化合物2,4,6,8,9,11-六叔丁氧羰基-3,7,10-三氧代-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HBOCP); 第二步HBOCP在三氟乙酸作用下脱掉保护基团叔丁氧羰基(BOC),减压蒸馏,过滤、洗涤,干燥后得到PTO,两步反应总收率为80.2%。
本文自行设计了PTO的合成路线及合成工艺,采用与羰基二咪唑一步合成PTO,具体合成工艺见2.2.3,合成路线见Scheme 4。
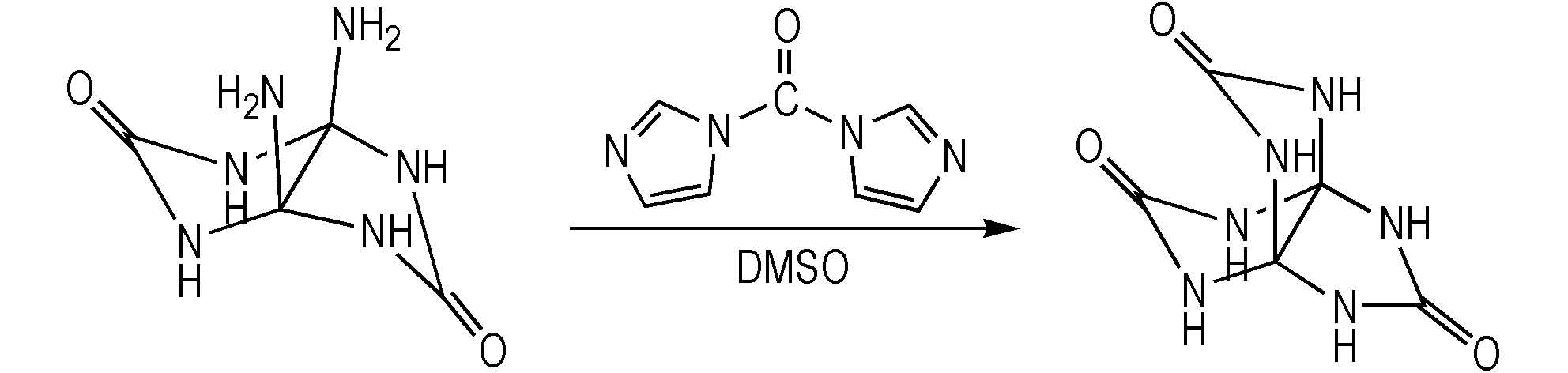
Scheme 4 One-step synthetic route of PTO
两种方法相比,本研究采用的PTO的合成方法反应步骤较少,总反应步骤为一步,反应收率为85.6%,比文献[13]两步总收率80.2%较高,相比参考文献[13]需要涉及萃取、蒸馏、色谱柱纯化、过滤、洗涤等操作过程,工艺复杂,而本方法仅需将反应液过滤、洗涤后即可得到PTO,操作工艺简单。
3.4.2 PTO纯化研究
PTO仅在DMSO中有一定的溶解度,但在DMSO中易形成过饱合态,难于重结晶,造成PTO的纯化有一定难度。本研究将PTO溶解在DMSO中,然后滴加到丙酮中,固体析出,过滤,滤饼用甲醇煮洗,过滤,干燥后纯度仅能达到93.5%( HPLC),故PTO的提纯尚需进一步研究。
3.5 中间体PTO纯度对合成 HBPTO的影响
反应条件同2.2.4,考察PTO纯度对HBPTO收率的影响,见结果见表4。
表4PTO纯度对HBPTO收率的影响
Table4EffectofpurityofPTOontheyieldofHBPTO

purity/%75.380.686.493.5yield/%//523.2purity/%//98.298.5
中间体PTO的纯度对HBPTO的收率影响较大,但PTO在大多数的溶剂中不溶,造成其提纯较为困难, 纯度为75.3%、80.6%的PTO样品均为前期获得的粗品纯度,没有提纯即投入下一步使用。86.4%、93.5%为采用不同提纯方法获得的PTO样品纯度,均为实测样品纯度。从表4可以看出,当PTO纯度小于80.6%,不能合成出HBPTO,这可能一方面是由于PTO的杂质中含有活泼氢,优先与PTO发生反应,随着纯度的提高,杂质对反应的影响降低,当PTO纯度为93.5%,收率达到23.2%。另一方面是由于PTO的6个N上的活泼H首先与NaH反应成盐后,原位进攻苄溴,发生烷基化反应,该反应具有反应位次较多,位阻较大的特点,因此造成该化合物的收率较低。
4 结 论
(1)首次在国内合成了3,7,10-三氧代-2,4,6,8,9,11-六苄基-2,4,6,8,9,11-六氮杂[3,3,3]螺桨烷(HBPTO),该化合物的合成以尿酸为起始原材料,经加成、氧化、缩合、取代反应等三步反应合成了含能材料中间体HBPTO,采用红外光谱、核磁共振、质谱以及元素分析等进行了结构表征,总收率为8.58%。
(2)探讨了尿酸合成DAGU反应机理,揭示了其氧化-加成微观反应过程。
(3)优化了氧化-加成反应合成DAGU条件,其最佳反应条件为:n(UC)∶n(K3Fe(CN)3)=1∶4.2,反应温度为25 ℃,反应时间为0.5 h。
(4)自行设计了二氨基甘脲与N,N′-羰基二咪唑一步缩合合成了PTO的新方法,与文献的两步法相比,具有反应步骤少、工艺简单的特点。
参考文献:
[1] Ibuka T, Inubushi Y, Tanaka K. Total Synthesis of the alkaloid hasubanonine[J].Chemical&PharmaceuticalBulletin, 1974, 22(4): 782-796.
[2] Zu L, Boal B W, Garg N K. Total synthesis of Aspidophyline A[J].JournaloftheAmericanChemicalSociety, 2011, 133: 8877-8879.
[3] Jian M H, Ritsuko Y, Chun S Y. Solid phase synthesis of 1,3-disubsituted succinimides[J].TetrahedronLetters, 2000, 41: 6111-6114.
[4] Sha C K, Wong D C. Intramolecular radical cyclization of silylacetylenic or olefinic α-iodo ketones application to the total synthesis of modhephene[J].TetrahedronLetters, 1990, 31(26): 3745-3748.
[5] Lee H Y, Kim S, Kim D I. Tandem radical cyclization reation of N-aziridinyl imines to [3,3,3]propellanes:formal total synthesis of di-modhephene[J].JournaloftheChemicalSociety, 1996, 61: 1539-1540.
[6] Nicolaou K C, Snyder S A, Montagnon T. The diels-alder reaction total synthesis[J].AngewandteChemieInternationalEdition, 2002, 41: 1668-1698.
[7] Trost B M, Shi Y. A Pd-Catalyzed zipper Reaction[J].JournaloftheAmericanChemicalSociety, 1991, 113(2): 701-703.
[8] Asahi K, Nishino H. Manganese(III)-based dioxapropellane synthesis using tricarbonyl compounds[J].Tetrahedron, 2008, 64: 1620-1634.
[9] ZHANG Qing-hua, ZHANG Jia-heng, QI Xiu-juan. Molecular design and property prediction of high density polynitro[3,3,3]-propellane-derivatized frameworks as potential high explosives[J].TheJournalofPhysicalChemistry, 2014, 118: 10857.
[10] 王伯周,李辉,李亚南, 等. 呋咱醚含能化合物研究进展[J]. 含能材料, 2012,20(4): 385-390.
WAGN Bo-zhou, LI Hui, LI Ya-nan, et al. Review on energetic compounds based on furoxanyl ether[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2012, 20(4): 385-390.
[11] 贾思媛, 张海昊, 王伯周, 等. 3,4- 双(3′-氨基呋咱-4′-基)呋咱合成与表征[J].含能材料, 2013, 21(3): 289-293.
JIA Si-yuan, ZHANG Hai-hao, WANG Bo-zhou, et al. Synthesis and characterization 3,4-bis(3′-aminofurazal-4′-yl)furazan[J].CheineseJournalofEnergeticMaterials(HannengCailiao), 2013, 21(3): 289-293.
[12] 王锡杰,王伯周,贾思媛, 等.三呋咱并氧杂环庚三烯的合成研究(TFO)[J]. 含能材料, 2012, 20(2): 258-259.
WANG Xi-jie,WANG Bo-zhou, JIA Si-yuan, et al. Trifurazanooxaheptatrien (TFO) synthesis[J].CheineseJournalofEnergeticMaterials(HannengCailiao), 2012, 20(2): 258-259.
[13] Young G K, Kyoo H C.Hexaaza[3,3,3]propellane compounds as key intermedlates for new molecular explosives and a method for preparing the same: US 8609861[P]. 2013.
猜你喜欢
中学化学(2022年5期)2022-06-17
陶瓷学报(2021年5期)2021-11-22
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
科学技术与工程(2021年8期)2021-04-22
理科考试研究·高中(2019年8期)2019-09-19
山东工业技术(2016年15期)2016-12-01
化学教学(2015年11期)2015-12-19
中国洗涤用品工业(2015年9期)2015-02-28
中国塑料(2014年10期)2014-10-17
应用化工(2014年1期)2014-08-16
