
高通量DNA测序数据的生物信息学方法
2016-04-10 08:51:26詹晓娟姚登举朱怀球
大数据 2016年2期
詹晓娟,姚登举,朱怀球
1. 黑龙江工程学院计算机科学与技术学院,黑龙江 哈尔滨 150050;
2. 哈尔滨理工大学软件学院,黑龙江 哈尔滨 150040;3. 北京大学生物医学工程系,北京 100871
◎研究
高通量DNA测序数据的生物信息学方法
詹晓娟1,姚登举2,朱怀球3
1. 黑龙江工程学院计算机科学与技术学院,黑龙江 哈尔滨 150050;
2. 哈尔滨理工大学软件学院,黑龙江 哈尔滨 150040;3. 北京大学生物医学工程系,北京 100871
高通量测序技术产生的DNA序列数据长度较短,而且数据量非常巨大。分析了高通量测序环境下大数据的挑战和机遇,总结并讨论了数据压缩、宏基因组数据序列拼接、宏基因组数据序列分析方面的算法和工具等研究成果。最后,展望了高通量测序下DNA短读序列数据研究的发展趋势。
高通量DNA测序;生物信息学;短读序列数据压缩;短读序列数据拼接;短读序列数据分析
1 引言
高通量测序技术又称“下一代”测序(next-generation sequencing, NGS)技术[1],可以一次性测定几十万甚至几百万条序列,是现今应用最广泛的测序技术。相对于传统的Sanger测序技术[2],NGS具有高速、高通量、低价格等优点[3]。高通量测序数据广泛应用于生物学、医学、遗传科学等诸多领域,具有重要研究价值。许多大型的科学研究项目,如千人基因组计划(1 000 genome project)、DNA元件百科全书(encyclopedia of DNA elements)计划、国际癌症基因组计划(international cancer genome project)等,正以前所未有的速度产生海量DNA序列。截至2014年2月,仅登录在美国GenBank数据库中的DNA序列数据就有十万亿碱基对,所有高通量测序下的DNA短读序列数据大小达到上千PB。随着测序技术的不断改善和测序成本的持续降低,每天都会有海量的DNA序列产生,使得生物数据量呈指数规模增长,平均约每14个月增加一倍。图1对高通量测序平台下的短读(short reads,以下简称reads)序列数据和其他大数据领域的原始数据增长方式进行了比较,阴影区预报了未来的增长趋势,从图11可以看出,高通量测序下的基因组序列数据即短读序列数据的增长远大于摩尔定律的增长速度。计算机是存储和处理DNA数据的主要工具,其微处理器性能和存储设备容量平均18~24个月翻一番,而DNA测序数据平均4~5个月就翻一番,DNA测序数据的增长速度已经远远超过了计算机微处理器和存储设备的增长速度。面对如此迅速增长的庞大的短读序列数据集,如何有效管理、分析、充分利用这些信息,已成为生物信息学发展亟需解决的问题[4]。

图1 不同种类数据的近似增长趋势
2 生物大数据带来的新挑战
随着高通量测序技术的发展,各种生物学数据呈现爆炸式增长,并且这一趋势将随着生物测序技术的发展而进一步增强。面对生命科学领域的大数据分析任务,多种不同维度的数据整合、多学科交叉的数据分析以及经典的数据挖掘算法都面临新的挑战。
2.1 多学科交叉的挑战
自从1990年人类基因组计划正式启动以来,20余年间,各种基因组、蛋白质组、转录组、宏基因组等国际生物学研究合作计划开始启动或已完成,目前国际上已经成立了多个大的跨国科研合作机构,生物信息领域的国际合作与交流也不断加强(见表11)。各种组学和生物信息学领域的国际化和跨学科间的专家合作使得团队成员在该领域取得了突出的成果,不仅发表了很多有影响力的文章,而且开发出许多新的数据集成和分析工具,以便资源和信息共享[5]。然而,面对飞速增长的生物学大数据和日渐增多的生物信息学研究任务,跨学科的国际合作仍面临巨大的挑战,例如不同的实验室和平台产生的大数据如何实现无障碍的共享和协作分析,不同组学产生的数据如何有效地进行集成、管理、维护和更新,如何开发新型的面向生物学大数据分析的算法和工具等。
2.2 数据和工具的整合问题
目前主流的高通量测序平台主要有Roche/454焦磷酸测序、Solexa/Illumina边合成边测序和ABI SOLiD连接测序。高通量测序技术的读长较短,但测序深度可以在一定程度上弥补读长较短带来的问题。其中,454测序平台读长最长有450~800 bp,适合对未知基因组从头测序;Solexa/Illumina测序读长比454测序平台短,但测序通量高、价位低,适合基因组重测序;SOLiD读长也较短,但测序精度高,特别适合SNP检测等。目前应用较普遍的是Illumina测序平台,约占现有测序工具数量的一半。

表1 生物大数据项目合作计划
不同的测序平台产生的数据格式各不相同,常用的文件格式有.bam、.csfasta、.fasta、.fastq、.gvf、.sam、.tar、.tiff、.var、.vcf等。现有的数据分析工具大多只能分析特定格式的数据,在实际的数据分析过程中往往需要把不同格式的数据进行标准化并重新整合,因此会浪费很多时间进行数据的预处理。例如,不同测序平台会产生不同品质和长度的高通量短读数据,由于没有统一的行业标准来描述高通量测序下的核苷酸序列和质量分数值,导致需要跨平台进行序列分析。因此,开发一组可以运行在不同计算平台下的互操作数据分析工具是一个具有挑战性的课题。
表22列出了目前高通量测序下各种组学所使用的工具和方法。随着这些多样的组学数据的整合,数据分析和解释的规模大大增加,这样就对基因组学和生命科学领域的大数据工具和基础设施提出更高的要求。对不同来源、不同形式的数据进行挖掘、评估、整合和应用还亟待加强。未来,多种组学数据的整合分析将会挑战传统的思维模式,发挥其至关重要的作用。
2.3 构建新型学术交流平台日益迫切
随着高通量测序成本的降低,生物大数据对于传统的数据存储、分析和解释提出了新的挑战,而将这些数据和成果进行系统整合并应用于医疗实践才刚刚开始。当前,一些小的实验室显然不具备存储和处理大数据的基础设施和能力。随着互联网技术的快速发展,众多的科学合作网络平台提供了实时的数据交换,使得人们可以通过互联网方便地进行数据分享和成果交流。例如,Illumina公司的新一代测序云计算平台BaseSpace(www.basepace. c o m)、开放科学框架平台(h t t p:// openscienceframework.org)和Figshare(http://epic.org/privacy/medical)等。全球三大IT公司Amazon、Rackspace和Google都提供了云存储和计算解决方案,通过云计算平台可以实现大型数据中心的资源共享。然而,云计算基因组学也面临着数据隐私和病人数据的合法性问题,拓展新型的学术交流平台成为生物大数据研究的一个重要任务。
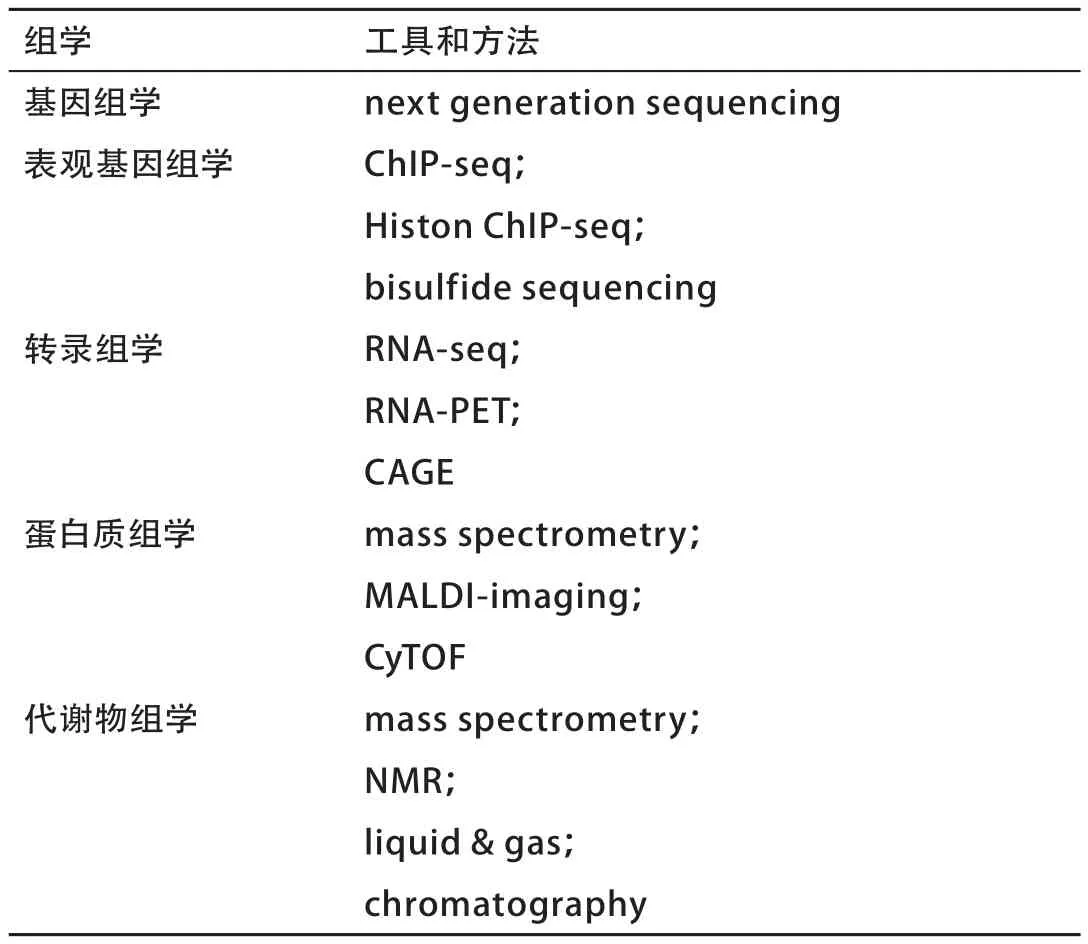
表2 高通量测序下各种组学所使用的技术
2.4 数据挖掘技术在生物大数据处理中的挑战
面对高通量测序数据的爆发式增长,传统的数据挖掘算法和工具遭遇巨大的挑战:如何建立智能学习数据库系统;如何对生物大数据存储访问和计算;如何进行隐私保护;如何结合领域知识设计新的适用于生物大数据挖掘分析的算法和工具。具体来说,面向生物学数据挖掘的数据挖掘技术主要有3个层次的挑战。第一个挑战是数据的访问和程序的运算。因为大数据都是分布式存储的,随着数据量的增长,如何建立一个有效的平台,使分散存储的数据能够摆脱计算机内存的限制和大数据处理的障碍,进行分布式计算。第二个挑战是不同的大数据有不同的语义和领域知识,如何能够更好地挖掘语义和领域知识,为数据所有者和消费者服务。第三个挑战集中在算法设计方面,生物大数据稀疏且具有各种各样的混合数据,数据有不确定性、不完整性和多源性等特点,如何用数据融合技术进行处理,并且挖掘出蕴含其中的复杂和动态信息;如何通过局部学习,得到一个反映全局问题的融合模型[17]。
3 高通量DNA测序数据的生物信息学方法
随着生物信息技术突飞猛进地发展,越来越多的计算机和数学领域的专家加入生物信息学研究的队伍,开发出许多好用的生物信息学工具,使得生物学、医学领域的专家可以利用这些先进工具对生物大数据进行分析,更准确地揭示生物进化的内部规律,更好地解释遗传变异,为基础医学研究向医学临床应用转化提供新思路和新方法,取得了非常有意义的成果。但是NGS测序的样本制备过程非常复杂,并且生成的序列难以处理,这给生物信息学专家带来了很大的挑战。
3.1 高通量DNA测序数据的压缩算法
NGS测序下的短读序列的数据量呈爆炸性增长,如果不对其进行压缩而直接存储或传输会消耗巨大的硬件存储设备,同时也会给网络传输带来很大的负担。NGS测序数据有其自身的特点和规律,存在大量的信息冗余,传统的数据压缩算法并不能够很好地压缩DNA序列,这就需要开发专门针对DNA序列的数据压缩算法和工具。
近几年,已经研发了许多专门针对NGS数据的压缩算法和工具,大多数是针对FASTQ格式的数据。根据DNA序列是否有参考基因组,压缩方法分为有参考基因组的压缩和无参考基因组的压缩。有参考基因组的数据压缩是利用参考基因组和短读序列的差异信息来进行压缩。这种方法第一步先把短读映射到参考基因组,记录每条短读在参考基因组上的位置以及与参考基因组的差异信息,然后再采用高效编码方式存储这些记录,实现数据压缩。其代表算法有DNAzip[18]、BWB[19]、SlimGene[20]、GRS[21]、mZIP[22]、NGC[23]、samcomp[24]等。由于同源物种基因组之间具有高度相似性,这种压缩通常能达到很高的压缩比,但这种方法有明显的局限性,有些测序数据(如宏基因数据、从头测序数据)并不存在现成的参考基因组,因此无法使用此算法;另外,该方法对于参考基因组依赖性太强,压缩和解压缩都需要相同参考基因组,这样参考基因组必须事先保存在本地,如果参考基因组缺失将直接影响压缩数据的使用。
无参考基因组的数据压缩方法通常采用两步法,首先最大限度地识别冗余DNA序列,然后再利用通用的压缩方法(如gzip、bzip2)进行处理。其代表算法工具有Beetl[25]、SCALCE[26]、SRComp[27]和ORCOM[28]。Beetl采用Burrows Wheeler变换算法,识别冗余;SCALCE采用局部一致性技术方法排序短读序列,识别关键子串;SRComp采用burstsort排序的方法,使相同的字符串聚集在一起,然后再采用不同的编码方式对其进行编码。ORCOM采用并行的Minimizers算法压缩reads中的重叠区域(overlap)。另一种新颖的无参考基因组的数据压缩方法是基于拼接的方法,代表算法有Quip[29]。Quip方法采用拼接的方式,用一小部分短读拼接成叠连群作为临时参考基因组,然后利用基于参考基因组的压缩方法进行压缩。
尽管高通量测序数据的压缩研究已取得一定成果,但其在计算资源、压缩算法方面仍面临巨大挑战。随着DNA测序数据量的增大,对计算资源的要求也越来越大,处理时间过长是DNA测序数据分析最重要的问题。另外,如何利用高通量测序技术产生有意义的冗余信息、采用并行化策略和基于索引的压缩方法、建立统一的数据质量评价标准等,都是重要的研究方向。
3.2 高通量DNA测序的序列拼接
由于测序技术的限制,新一代测序的读长较短(30~500 bp)[30],测序所得序列无法满足大多数序列分析的需要[31],因此序列拼接成为基因组学研究中一个重要的环节。所谓序列拼接,是指将测序得到的短序列片段利用计算的方法拼接成较长的连续序列片段(contig)或者中间带有空隙的长序列片段(scaffold)乃至整段基因组序列的方法。
序列拼接包括两种不同的策略:从头(D e N o v o)拼接的方法和对照(comparative)拼接的方法[32]。从头拼接是指在没有任何基因组序列参照的前提下,构建全新基因组序列的策略,而对照拼接是指在参照基因组序列的指导下进行的基因组序列的拼接。对照拼接适用于存在参照基因组序列的拼接,比如重测序项目中的序列拼接,而对于全新物种的大规模全基因组测序以及宏基因组测序项目主要使用从头拼接。
拼接算法的主要挑战来源于基因组中的重复序列片段。在不同区域的两个完全一致的重复片段无法通过计算的方式来辨别。对于相似但不完全一致的重复片段,可以通过提高序列比对的相似度阈值区分不同的复本,这种方法一般还涉及对reads中测序错误的估计[33]。重复片段的区分一般需要借助于reads或是mate-pair的跨越。所谓的mate-pair是指测序时从一段长度已知的片段两端测得的一对reads。对于reads来说,如果reads的中间是重复序列,而两端都有足够长的唯一片段,则可以区分中间的重复片段,这种方法针对短的重复片段有效,一般在k-mer图算法中使用。对于mate-pair来说,如果matepair分别处于重复序列的两端,也可以指导正确的拼接路径,而且mate-pair比reads更长,因此可以区分更长的重复片段。高的测序深度有利于重复片段的区分,因为高的测序深度可能提供更多的reads或者mate-pair跨越重复片段。对于新一代测序中短序列的拼接,重复片段的区分更加困难,因为reads更短,更多的重复片段无法通过reads来区分,因此提高测序深度和使用mate-pair尤为重要。
测序错误也给重复片段问题的解决增加了难度。因为拼接算法必须因为测序错误而接受不完全一致的重叠,以免错漏了真实的重叠。然而对测序错误的容忍又增加了拼接的假阳性。更多不完全一致的重复片段会对算法造成麻烦。另外,序列拼接需要考虑的一个问题是计算时间上的复杂度问题,尤其对于reads数量越来越多的大规模测序数据。例如,为了提高拼接效率,所有的拼接软件都在不同程度地以不同方式使用k-mer的概念。很直观的一个结论是,reads之间的重叠区域必然共同享有k-mer。而对共享k-mer的搜索显然要比计算序列比对简单得多。因此,几乎所有的拼接算法都涉及对k-mer的计算。
理论上,序列拼接属于一个NP难的问题,尚无一个盖棺定论的解答方法。现有的拼接算法只能通过一系列复杂的推断性质的步骤来获得近似的“解答”。这些算法仍有局限性,例如拼接结果错误、拼接序列连续性差、计算时间长、内存消耗量大等。因此,序列拼接算法仍有很大的改进空间。另外,测序技术的不断变化和改进,使得新数据对序列拼接不断提出新的要求,以更好地适应新数据的特点。
3.3 高通量测序下宏基因组的基因预测方法
基于高通量测序的宏基因组学研究给环境相关微生物的研究带来了新的机遇。随着越来越多的各种生态环境中宏基因组序列被测定并公开,有效的宏基因组数据分析和功能预测软件被开发与应用,这些都大大推动了宏基因组学的发展。目前研究基因预测的方法主要有两类:一类是基于序列相似性的预测方法,基于已知的基因序列通过搜索相似度较高的序列进行预测;另一类是基于统计学模型的预测方法,即利用数学统计模型进行基因预测,从已知的DNA序列中训练出统计学模型,应用到宏基因组的测序结果上进行预测。
(1)基于序列相似性比较的方法
序列比对是生物信息学的基础,其基本问题是比较两个或两个以上序列之间的相似性。两个序列比对已有发展成熟的动态规划(dynamic programming)算法和在此基础上发展起来的工具包BLAST[34]和FASTA[35]。事实上,在基于比对的方法中,高通量测序所得的序列较短,而这种短序列直接进行比对的效果往往不理想,并且大量的原始数据进行比对会耗费很多时间,因此需要在比对前进行序列拼接,将其拼接成较长的序列,提高分析效率和分析效果[36]。由于必须与已知基因序列进行相似性比较,故这种方法很难发现新基因。
基于序列相似性比较的高通量测序的宏基因组数据的应用非常多。2010年,华大基因在Nature发表文章,对人体肠道微生物基因组研究计划(MetaHIT)进行了总结[37]。该计划为研究人体肠道微生物群落与人类健康之间的关系,采集了124个欧洲人的粪便样本,其中包括25个炎症性肠病(inflammatory bowel disease,IBD)患者和99个健康志愿者的样本,并用Illumina测序平台进行测序,产生了567.7 GB的测序数据,并对序列拼接、注释、功能基因的分类、多态性分析等进行了研究。2012年,华大基因在Nature发表了一篇研究人体肠道微生物与Ⅱ型糖尿病之间关系的文章[38]。该研究收集了345个中国人的肠道微生物样本,用Illumina测序平台对其进行了深度测序,并在基因组关联研究(genome wide association studies,GWAS)的基础上开发了一种全基因组相关联研究(metagenome wide association studies,MGWAS)的方法,对Ⅱ型糖尿病与肠道微生物失调之间的关系进行了深入研究。人体肠道中绝大多数种类的微生物是难以培养的,只有运用宏基因组学技术才能研究人类肠道中的所有微生物群落,进而了解人类肠道中细菌的物种分布。
(2)基于序列内容统计特征的方法
基于序列内容统计特征的基因预测方法一般是建立在密码子的编码区和非编码区有不同相对出现频率的基础上的。除了一个区域碱基组成的特征外,基因长度分布、CG含量、基因重叠区域的特征等因素也常被用于基因预测中。根据DNA序列中编码蛋白质区域和非编码区域内容统计特征的差别,建立其学习模型,可以有效地进行基因预测。在单个基因组上具有代表性的方法包括采用马尔科夫模型的GeneMark[39-41]系列、Glimmer[42,43]系列、FGENESB[44]和MED[45,46]系列。GeneMark对原核生物、真核生物和病毒均能进行基因预测。Glimmer被广泛应用于微生物的基因预测。FGENESB主要用于细菌基因组的基因自动预测和注释。MED是笔者所在课题组开发的一款基于多元熵距离法的原核生物基因预测算法,该算法的基础为开放阅读框(ORF)和翻译起始位点(TIS)的综合统计模型。MED2.0在对DNA的GC核苷酸含量高的细菌基因组和古细菌基因组的基因预测上具有明显优势,之后又推出了MED2.1,提高了预测精度,达到了国际水平。
针对宏基因组序列的研究,研究人员开发了一系列宏基因组预测算法(见表33)。宏基因组预测算法借鉴了传统的基于单基因组的基因预测方法,只是对原始数据增加了预处理的步骤。例如,MetaGUN算法基于序列组成的统计特征对输入序列进行分类,对同一类中的序列使用相同的统计模型刻画,然后分别独立地进行基因预测,在模拟宏基因序列测试集和在两个人体肠道微生物的真实数据上的测试表明,MetaGUN在发现新基因方面更具潜力。MetaGeneMark同时使用细菌—古细菌和嗜温细菌—嗜热细菌两套模型进行预测。FragGeneScan适用于有测序错误的宏基因组序列。
近年来,专门针对宏基因组序列的基因预测方法目前面临着新的挑战,基于序列相似性比较的方法,使用BLAST系统工具对已知数据库进行相似性搜索,依赖性强,无法发现新基因。基于统计建模的预测算法运行速度快,在保证高特异性的条件下能获得更高的敏感性。宏基因组序列来源于繁杂且大多为未知的物种,微生物中已知的细菌和古细菌只占全世界存在量的10%;同时高通量测序的宏基因组DNA序列很短,存在大量不完整基因,无法在单个序列片断上完成自学习,为统计建模所能提供的信息有限;另外,如何把分析结果和已知的数据库(Greengenes[55]、SILVA[56]等)结合起来、如何进一步研究生物体之间以及生物体和环境之间的相互作用等,都成为亟待解决的问题。

表3 宏基因组基因预测算法
4 结束语
高通量测序技术奠定了生物信息学的“大数据”基础,面对如潮水般的基因序列数据,给后续基因组分析方法的研究和工具的发展带来了巨大挑战。本文总结讨论了高通量测序数据的基因组分析及生物信息学方法。目前,基因组生物信息学研究正面临从传统的全基因组序列分析到当前基于短读的序列片段(含contigs)分析;从传统的单个物种的全基因组序列分析到当前多个物种混杂的序列片段数据集的分析;从本地计算机运算分析到未来适应“云计算”模式的远程、快速运算分析这几方面发展。面对如此快速的发展,现有的生物信息学方法和工具已经不能满足如此大量的数据资料的需求,只有进一步发展出优秀的生物信息学方法和工具,才能更好地利用高通量测序技术的优势和应用价值。
[1] S C H US T ER S C. N e x t-g e n e r a t i o n sequencing transforms today’s biology[J]. Nature Methods, 2008, 5(1): 16-18.
[2] SANGER F, NICKLEN S, COULSON A R. DNA sequencing with chain-terminating inhibitors[J]. Proceeding of the National Academy of Sciences, 1977, B7(12): 5463-5467.
[3] SHENDURE J, JI H. Next-generation DNA sequencing[J]. Nature Biotechnology, 2008, 26(10): 1135-1145.
[4] HIGGINS G. Human Genomes and Big Data Challenges[R]. Mason: AssureRx Health Inc, 2013.
[5] WARD R M, SCHMIEDER R, HIGHNAM G, et al. Big data challenges and opportunities in highthrough-put sequencing[J]. Systems Biomedicine, 2013, 1(1): 29-34.
[6] DUNHAM I, BIRNEY E, LA JOIE B R, et al. An integrated encyclopedia of DNA elements in the human genome[J]. Nature, 2012, 489(7414): 57-74.
[7] COLLINS F S, BARKER A D. Mapping the cancer genome[J]. Scientific American, 2007, 296(3): 50-57.
[8] HAYDEN E C. International genome project launched[J]. Nature, 2008, 451(7177): 378-389.
[9] GEVERS D, KNIGHT R, PETROSINO J F, et al. The human microbiome project: a community resource for the healthy human microbiome[J]. PLoS Biology, 2012, 10(8): e1001377.
[10] HAUSSLER D, O’BRIEN S J, RYDER O A, et al. Genome 10K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species[J]. The Journal of Heredity, 2008, 100(6): 659-674.
[11] O’ROAK B J, VIVES L, GIRIRAJAN S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations[J]. Nature, 2012, 485(7397): 246-250.
[12] EHRLICH S D. MetaHIT: the European union project on metagenomics of the human intestinal tract[M]// Metagenomics of the Human Body. New York: Springer, 2011: 307-316.
[13] L E G R A I N P, A E B E R S O L D R, A R C H A KO V A, e t a l. T h e h u m a n proteome project: current state and future direction[J]. Molecular & Cellular Proteomics, 2011, 10(7): M111. 009993.
[14] G I L B E R T J A, M E Y E R F, ANTONOPOULOS D, et al. Meeting report: the terabase metagenomics workshop and the vision of an earth microbiome project[J]. Standards in Genomic Sciences, 2010, 3(3): 243.
[15] R O B I N S O N G E, H A C K E T T K J, PURCELL M M, et al. Creating a buzz about insect genomes[J]. Science, 2011, 331(6023): 1386.
[16] JOLY Y, DOVE E S, KNOPPERS B M, et al. Data sharing in the post-genomic world: the experience of the international cancer genome consortium (ICGC) data access compliance office (DACO)[J]. PLoS Comput Biol, 2012, 8(7): e1002549.
[17] WU X D, ZHU X Q. Data mining with big data[J]. IEEE Transactions on Knowledge and Data Engineering, 2014, 26(1): 97-108.
[18] C H R I S T L E Y S, L U Y, L I C, e t a l. Human genomes as email attachments[J]. Bioinformatics, 2009, 25(2): 274-275.
[19] BRADON M C, WALLACE D C, BALDI P. Data structures and compression algorithms for genomic sequence data[J]. Bioinformatics, 2009, 25(14): 1731-1738.
[20] K O Z A N I T I S C, S A U N D E R S C, K RUGLYAK S, e t al. Co m p r e ssin g genomic sequence fragments using SlimGene[J]. Journal of Computational Biology, 2011, 18(3): 401-413.
[21] WANG C, ZHANG D. A novel compression tool for efficient storage of genome resequencing data[J]. Nucleic Acids Research, 2011, 39(7): e45.
[22] F R I T Z M H Y, L E I N O N E N R, COCHRANE G, et al. Efficient storage of high throughput DNA sequencing data using reference-based compression[J]. Genome Research, 2011, 21(5): 734-740.
[23] MILLER J R, KOREN S, SUT TON G. Assembly algorithms for next-generation sequencing data[J]. Genomics, 2010, 95(6): 315-327.
[24] B O N F I E L D J K, M A H O N E Y M V. Compression of FASTQ and SAM format sequencing data[J]. Plos One, 2013, 8(3): 1453-1456.
[25] COX A J, BAUER M J, JAKOBI T, et al. Large-scale compression of genomic sequence databases with the Burrows-Wheeler transform[J]. Bioinformatics, 2012, 28(11): 1415-1419.
[26] H A C H F, N U M A N A G I Ć I, A L K A N C, et al. SCALCE: boosting sequence compression algorithms using locally consistent encoding[J]. Bioinformatics, 2012, 28(23): 3051-3057.
[27] SELVA J J, CHEN X. SRComp: short read sequence compression using burstsort and Elias omega coding[J]. PloS One, 2013, 8(12): e81414.
[28] P AT R O R, K I N G S F O R D C. D a t adependent bucketing improves referencefree compression of sequencing reads[J]. Bioinformatics, 2015: btv248.
[29] J O N E S D C, RUZ ZO W L, PEN G X, et al. Compression of next-generation sequencing reads aided by highly efficient de novo assembly[J]. Nucleic Acids Research, 2012, 40(22): e171.
[30] METZKER M L. Applications of nextgeneration sequencing technologies the next generation[J]. Nature Reviews Genetics, 2010, 11(1): 31-46.
[31] WOOLEY C, GODZIK A, FRIEDBERG I. A primer on metagenomics[J]. PLoS Comput Biol, 2010, 6(2): e1000667.
[32] POP M, PHILLIPPY A, DELCHER A L, et al. Comparative genome assembly[J]. Briefings in Bioinformatics, 2004, 5(3): 237-248.
[33] KECECIOGLU J, JU J. Separating repeats in DNA sequence assembly[C]// The 5th Annual International Conference on Computational Biology, April 22-25,2001, Montreal, Canada. [S.l.:s.n.], 2001: 176-183.
[34] P R I D E D T, M E I N E R S M A N N R J, WASSENAAR T M, et al. Evolutionary implications of microbial genome tetranucleotide frequency biases[J]. Genome Research, 2003, 13(2): 145-158.
[35] WU Y W, YE Y. A novel abundance-based algorithm for binning metagenomic sequences using l-tuples[J]. Journal of Computational Biology, 2011, 18(3): 523-534.
[36] PRAKASH T, TAYLOR T D. Functional assignment of metagenomic data: challenges and applications[J]. Briefings in Bioinformatics, 2012, 13(6): 711-727.
[37] QIN J, LI R, RAES J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464(7285): 59-65.
[38] QIN J, LI Y, CAI Z, et al. A metagenomewide association study of gut microbiota in type 2 diabetes[J]. Nature, 2012, 490(7418): 55-60.
[39] B O R O D O V S K Y M, M C I N I N C H J. GENMARK: parallel gene recognition for both DNA strands[J]. Computers & Chemistry, 1993, 17(2): 123-133.
[40] L U K A S H I N A, B O R O D O V S K Y M. GeneMark.hmm: new solutions for gene finding[J]. Nucleic Acids Research, 1998, 26(4): 1107-1115.
[41] B E S E M E R J, L O M S A D Z E A, BORODOVSKY M. GeneMarks: a selftraining method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions[J]. Nucleic Acids Research, 2001, 29(12): 2607-2618.
[42] SALZBERG S L, DELCHER A L, KASIF S, et al. Microbial gene identification using interpolated Markov models[J]. Nucleic Acids Research, 1998, 26(2): 544-548.
[43] DELCHER A L, BRATKE K A, POWERS E C, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer[J]. Bioinformatics, 2007, 23(6): 673-679.
[44] FRIGAARD N U, MARTIMEZ A, MINCER T J, et al. Proteorhodopsin lateral gene transfer between marine planktonic bacteria and archaea[J]. Nature, 2006, 439(7078): 847-850.
[45] OUYANG Z, ZHU H, WANG J, et al. Multivariate entropy distance method for prokaryotic gene identification[J]. Journal of Bioinformatics and Computational Biology, 2004, 2(2): 353-373.
[46] ZHU H Q, HU G Q, YANG Y F, et al. MED: a new non-super vised gene prediction algorithm for bacterial and archaeal genomes[J]. BMC Bioinformatics, 2007, 8(1): 97.
[47] NOGUCHI H, TANIGUCHI T, ITOH T. MetaGeneAnnotator: detecting speciesspecific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes[J]. DNA Research, 2008, 15(6): 387-396.
[48] H O F F K J, L I N G N E R T, M E I N I C K E P, et al. Orphelia: predicting genes in metagenomic sequencing reads[J]. Nucleic Acids Research, 2009, 37(suppl 2): W101-W105.
[49] ZHU W, LOMSADZE A, BORODOVSKY M. Ab initio gene identification in metagenomic sequences[J]. Nucleic Acids Research, 2010, 38(12): e132.
[50] RHO M, TANG H, YE Y. FragGeneScan: predicting genes in short and error-prone reads[J]. Nucleic Acids Research, 2010, 38(20): e191.
[51] KELLE Y D R, LIU B, DELCHER A L, et al. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering[J]. Nucleic Acids Research, 2012, 40(1): e9.
[52] HYATT D, LOCASCIO P F, HAUSER L J,et al. Gene and translation initiation site prediction in metagenomic sequences[J]. Bioinformatics, 2012, 28(17): 2223-2230.
[53] WANG Y, LEUNG H C M, YIU S M, et al. MetaCluster 5.0: a two-round binning approach for metagenomic data for lowabundance species in a noisy sample[J]. Bioinformatics, 2012, 28(18): i356-i362.
[54] L I U Y, G U O J, H U G, e t a l. G e n e prediction in metagenomic fragments based on the SVM algorithm[J]. BMC Bioinformatics, 2013, 14(suppl 5): S12.
[55] D E S A N T I S T Z, H U G E N H O LT Z P, L A R S E N N, e t a l. G r e e n g e n e s, a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and Environmental Microbiology, 2006, 72(7): 5069-5072.
[56] PRUESSE E, QUA ST C, K NIT TEL K, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB[J]. Nucleic Acids Research, 2007, 35(21): 7188-7196.
Bioinformatics methods for high-throughput DNA sequencing data
ZHAN Xiaojuan1, YAO Dengju2, ZHU Huaiqiu3
1. College of Computer Science and Technology, Heilongjiang Institute of Technology, Harbin 150050, China
2. School of Software, Harbin University of Science and Technology, Harbin 150040, China 3. Department of Biomedical Engineering, Peking University, Beijing 100871, China
DNA sequence data generated by high-throughput sequencing technology is short in length, and the amount of data is enormous. The challenges and opportunities of the big data in high-throughput sequencing environment were analyzed. The data compression, the assembly of metagenomic sequence data, and algorithms and tools of metagenomic sequence data analysis also were summarized and discussed. Finally, the future of the study on short read DNA sequence data in high-throughput sequencing environment was discussed.
high-throughput DNA sequencing, bioinformatics, short read sequence data compression, short read sequence data splicing, short read sequence data analysis
TP399
A
10.11959/j.issn.2096-0271.2016021

詹晓娟(1978-),女,黑龙江工程学院讲师,主要研究方向为数据挖掘、机器学习、生物信息。

姚登举(1980-),男,哈尔滨理工大学副教授,主要研究方向为数据挖掘、机器学习、生物信息。

朱怀球(1970-),男,北京大学教授,主要研究方向为生物医学信息学和计算系统生物学。
2015-09-30
黑龙江省自然科学基金资助项目(No.F201313);黑龙江省教育厅科学技术研究资助项目(No.12541124);哈尔滨市科技创新人才资助项目(No.2013RFQXJ114)
Foundation Items: The Natural Science Foundation of Heilongjiang Province (No.F201313), The Foundation of Heilongjiang Province Educational Committee (No.12541124), The Harbin Special Funds for Technological Innovation Research of Heilongjiang Province of China (No.2013RFQXJ114)
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
今日农业(2021年11期)2021-08-13 08:53:24
现代畜牧科技(2021年4期)2021-07-21 06:12:50
透析与人工器官(2020年1期)2020-11-16 01:42:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
中国博物馆(2018年2期)2018-12-05 05:28:50
中国发展观察(2017年8期)2017-04-26 03:51:50
现代检验医学杂志(2015年4期)2015-02-06 02:01:55
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
