
甘草次酸微乳的制备及质量评价
2016-04-05 06:59:17肖志偲赵瑞芝卢传坚
中成药 2016年1期
肖志偲, 赵瑞芝, 卢传坚
(广州中医药大学第二临床医学院,广东广州510006)
甘草次酸微乳的制备及质量评价
肖志偲, 赵瑞芝*, 卢传坚
(广州中医药大学第二临床医学院,广东广州510006)
摘要:目的 制备甘草次酸微乳,并对其性质进行考察。方法 HPLC测定甘草次酸的含有量;微孔酶标测定浊度法绘制伪三元相图;星点设计-效应面法优选微乳处方。结果 甘草次酸微乳的最优处方为聚氧乙烯40氢化蓖麻油(RH40)∶二乙二醇单乙基醚(Transcuto1P)∶油酸聚乙二醇甘油酯(Labrafi1M 1944 Cs)∶纯水=6.76∶2.25∶1∶14.37(质量比);微乳含药量(3.00±0.06)mg/g;外观澄清、透明且稳定;平均粒径(18.8±0.9)nm;多分散系数0.28±0.01;30℃恒温下黏度(0.089±0.003)Pa.s;28℃下pH值5.75±0.10,15 000 r/min离心15 min后无分层现象。结论 该微乳制剂稳定,可大幅提高甘草次酸的溶解度并显著增强其生物利用度,有望作为甘草次酸的新型给药制剂。
关键词:甘草次酸;微乳;质量评价;HPLC;微孔酶标;星点设计-效应面法
甘草次酸(G1ycyrrhetinic acid,GA)是甘草中主要的三萜类活性成分甘草酸的苷元,具有抗炎、抗病毒、抗肿瘤和防癌作用[1],由于其可抑制11β-羟基甾体脱氢酶的活性,也常用于银屑病等皮肤疾病的治疗[2-3]。然而,甘草次酸在水中的溶解性极差[4],导致其生物利用度较低,从而限制了其临床应用。
微乳(micro emu1sion,ME)是由油相、水相、表面活性剂和助表面活性剂通过适宜的配比,混合形成的粒径小于100 nm、呈透明或半透明、低黏度、热力学稳定且各向同性的胶体分散体系[5-7]。与常规制剂相比,微乳可提高难溶性药物在水中的溶解性,促进药物吸收,提高生物利用度。
本实验制备了甘草次酸微乳,并对其理化性质及含有量进行初步考察,期望利用新剂型来改善其溶解度和吸收特性,为将其开发成治疗银屑病的外用制剂奠定基础。
1 材料与仪器
1.1 试验材料 甘草次酸对照品(中国食品药品检定研究院,批号110723200612,供含有量测定用);甘草次酸(南京泽朗医药科技有限公司);油酸聚乙二醇甘油酯(Labrafi1M 1944Cs,法国Gattefosse公司);肉豆蔻酸异丙酯(IPM)、棕榈酸异丙酯(IPP)(纯度均>98%,阿拉丁试剂上海有限公司);聚山梨酯80(Tween 80,天津富宇精细化工有限公司,分析纯);聚氧乙烯35蓖麻油(EL-35)、聚氧乙烯40氢化蓖麻油(RH-40)(德国BASF公司);聚乙二醇400(PEG400,天津大茂化学试剂厂,分析纯);二乙二醇单乙基醚(Transcuto1P,法国Gattefosse公司)。甲醇为色谱纯(天津四友精细化学品有限公司);丙二醇为分析纯(天津百世化工有限公司);亚甲基蓝、苏丹红Ⅲ(天津市天新精细化工开发中心);其他试剂均为分析纯。
1.2 试验仪器 Agi1ent 1200高效液相色谱仪;恒温摇床(E25);高速离心机(5430R);涡旋混合仪(IKA);超声仪(USC-702);电热恒温水浴锅(DK-S26);全波长酶标仪(EonC);多功能搅拌机(IKA);黏度计(NDJ-1);pH酸度计(De1ta-312 pH计);电子天平(HTP-312);Zeta-粒度测定仪(ZETA-1000HSA)。
2 方法
2.1 甘草次酸微乳处方确定
2.1.1 溶解度实验 将过量的甘草次酸分别加入到含有不同油相、表面活性剂、助表面活性剂的离心管中,37℃恒温摇床中水平振摇24 h,4 000 r/min离心10 min,取上清液,适量甲醇稀释,高效液相色谱法测定其浓度。
甘草次酸测定参考文献[8]。采用Agi1ent C18色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-0.1%甲酸溶液,梯度洗脱(0~5 min,85∶15;5~12 min,100∶0);体积流量为1.0 mL/min;检测波长为248 nm;柱温为27℃;进样量为5 μL。
2.1.2 表面活性剂和助表面活性剂的选择 参照文献[9-10],将油相和混合表面活性剂按9∶1、8∶2、7∶3、6∶4、5∶5、4∶6、3∶7、2∶8、1∶9的比例(质量比)混合均匀成混合相。将各比例混合相按照A1200 μL、A2190 μL、A3180 μL至B810 μL加入96孔酶标板孔中,然后将水相按A210 μL、A320 μL至B8190 μL、B9200 μL加入酶标板孔中。涡旋震荡10 min后,于酶标仪630 nm波长下测定浊度,得到由澄清到混浊或者由混浊到澄清的临界点体系。然后,根据临界点绘制伪三元相图,由微乳区域的面积大小来确定表面活性剂和助表面活性剂[11]。
2.1.3 Km值的选择 将“2.1.2”项下的表面活性剂和助表面活性剂按照不同比例(即Km值)混合,加入一定量油相,按照“2.1.2”项下操作方法,根据临界点绘制伪三元相图,根据微乳区域大小确定Km值。
2.2 星点设计处方优化 以含水量(X1)及油相与混合表面活性剂的比值(O/S,X2)为自变量,选择含药量(Y1)、粒径(Y2)、黏度(Y3)和浊度(Y4)作为因变量,采用2因素5水平进行星点设计优化[12-14]。根据预实验,确定X1和X2极值范围分别为46%~74%和0.11~0.67。各因素水平代码值和实际值见表1。
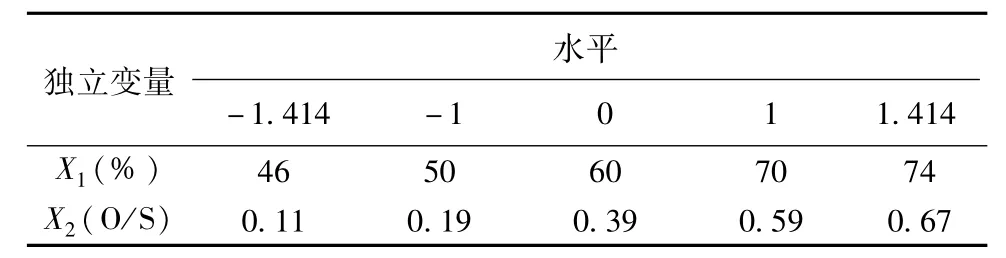
表1 各因素水平代码值和实际值
2.3 微乳含药量测定方法 精密移取微乳1 mL至10 m L量瓶中,加3~5 mL甲醇超声1 min进行破乳,然后甲醇定容至10 mL,摇匀。取2 mL至4 mL离心管中,12 000 r/min离心10 min。移取1 mL,0.45 μm滤膜过滤后,转移至进样瓶中,HPLC对样品进行含有量测定,平行操作3次,测定条件按照“2.1.1”项。
2.4 微乳理化性质的考察
2.4.1 微乳类型鉴别 取等量制备的含药微乳2份,分别加入等量红色油溶性染料苏丹红或蓝色水溶性染料亚甲蓝,观察两者在微乳中的扩散速度。
2.4.2 微乳黏度测定 取3批甘草次酸微乳样品各一份,采用NDJ-1型旋转黏度计在恒温下进行黏度测定。
2.4.3 粒径测定 取3批甘草次酸微乳样品各一份,室温条件下用水稀释10倍后,激光粒度分析仪测定其粒径。
2.4.4 稳定性初步考察 将制得的甘草次酸微乳以15 000 r/min离心15 min,观察有无分层现象。另取甘草次酸微乳3批,分别封存于5 mL西林瓶中,在相对湿度75%、温度25℃条件下,放置0、1、3个月,观察微乳是否保持澄清透明,有无油水分层现象。
3 结果
3.1 方法学考察结果 以色谱峰面积(Y)对甘草次酸的质量浓度(X)进行线性回归,得回归方程为Y=7.091 1X+ 11.258,r2=0.999 6,线性范围为62.5~1 000 μg/mL。高、中、低浓度溶液日内精密度RSD为0.15%、0.53%、0.33%;日间精密度RSD为0.05%、0.44%、0.78%。平均加样回收率为98.2%,RSD值为1.68%。
3.2 处方筛选
3.2.1 油相的选择 甘草次酸在各种辅料中的溶解度见图1。由图可知,甘草次酸在Labrafi1M 1944Cs中的溶解度最大,故为了提高微乳的载药量,确定油相为Labrafi1 M 1944Cs。
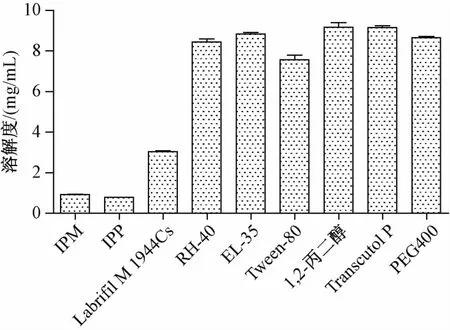
图1 甘草次酸在不同辅料中的溶解度(n=3)
3.2.2 表面活性剂和助表面活性剂的选择 以油酸聚乙二醇甘油酯为油相,聚氧乙烯40氢化蓖麻油、聚氧乙烯35蓖麻油为表面活性剂,1,2-丙二醇、聚乙二醇400、二乙二醇单乙基醚为助表面活性剂,分别绘制伪三元相图,见图2。由图可知,聚氧乙烯40氢化蓖麻油-二乙二醇单乙基醚组合的成乳区域最大,因此表面活性剂选择聚氧乙烯40氢化蓖麻油,助表面活性剂选择二乙二醇单乙基醚。
3.2.3 Km值的选择 以油酸聚乙二醇甘油酯为油相,聚氧乙烯40氢化蓖麻油为表面活性剂,二乙二醇单乙基醚为助表面活性剂,在表面活性剂和助表面活性剂的质量比分别为2∶1、3∶1、4∶1时绘制伪三元相图,见图3。由图可知,当Km为3时,微乳区域较大,故选择表面活性剂:助表面活性剂=3∶1进行处方优化。
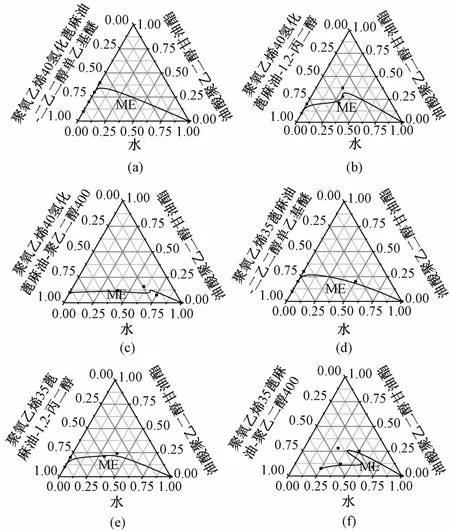
a.聚氧乙烯40氢化蓖麻油-二乙二醇单乙基醚 b.聚氧乙烯40氢化蓖麻油-1,2-丙二醇 c.聚氧乙烯40氢化蓖麻油-聚乙二醇400 d.聚氧乙烯35蓖麻油-二乙二醇单乙基醚 e.聚氧乙烯35蓖麻油-1,2-丙二醇 f.聚氧乙烯35蓖麻油-聚乙二醇400图2 不同表面活性剂和助表面活性剂的伪三元相图
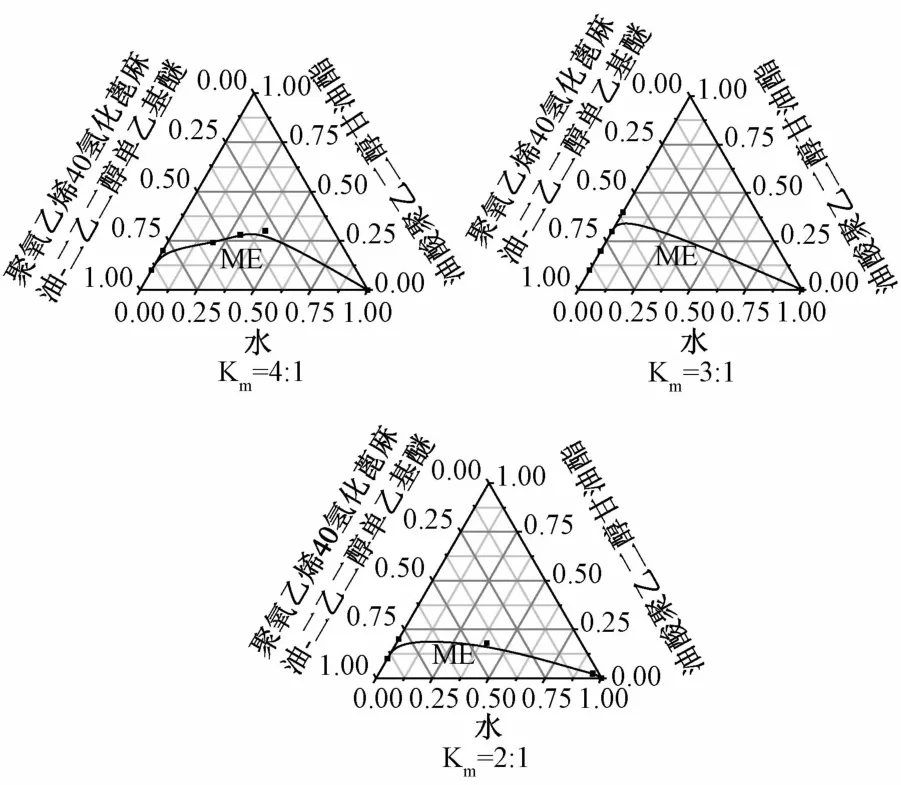
图3 不同Km值下的伪三元相图
3.3 甘草次酸微乳处方优化 以水相百分含有量X1(%)、油相/混合表面活性剂(O/S)X2为自变量,微乳含药量Y1、粒径Y2、黏度Y3、浊度值Y4为因变量,利用Design-expert8.0.6软件得到不同的处方。再称取处方量混合相成分,加入适量药粉,溶解至肉眼看不见药粉,再加入水相搅拌成乳。对各组合乳剂进行含量、粒径、黏度以及浊度测定,平行操作3次。结果见表2。
经线性及非线性回归处理后,粒径、浊度与O/S呈多元线性关系,随着O/S值增大而增大,而且受含水量影响较小;黏度与O/S呈非线性关系,含水量对其影响较大,在大于60%时变化较小。含药量符合二次多项式模型,进行二项式拟合(r2=0.998 9,P=0.000 3),得方程Y1= +9.313 45 -0.111 52X1(%)-4.769 66X2(O/S)+ 0.045 000X1(%)X2(O/S)+1.812 50×10-4X21(%)+1.265 63X22(O/S)。经优化,水相百分含有量X1= 58.93%,油相和混合表面活性剂的比例(O/S)X2= 0.111,预测值载药量3.04 mg/g,粒径20.6 nm,黏度0.1 Pa.s,浊度值0.04。因此,甘草次酸微乳的最优处方为聚氧乙烯40氢化蓖麻油∶二乙二醇单乙基醚∶油酸聚乙二醇甘油酯∶纯水的比例为6.76∶2.25∶1∶14.37(质量比)。
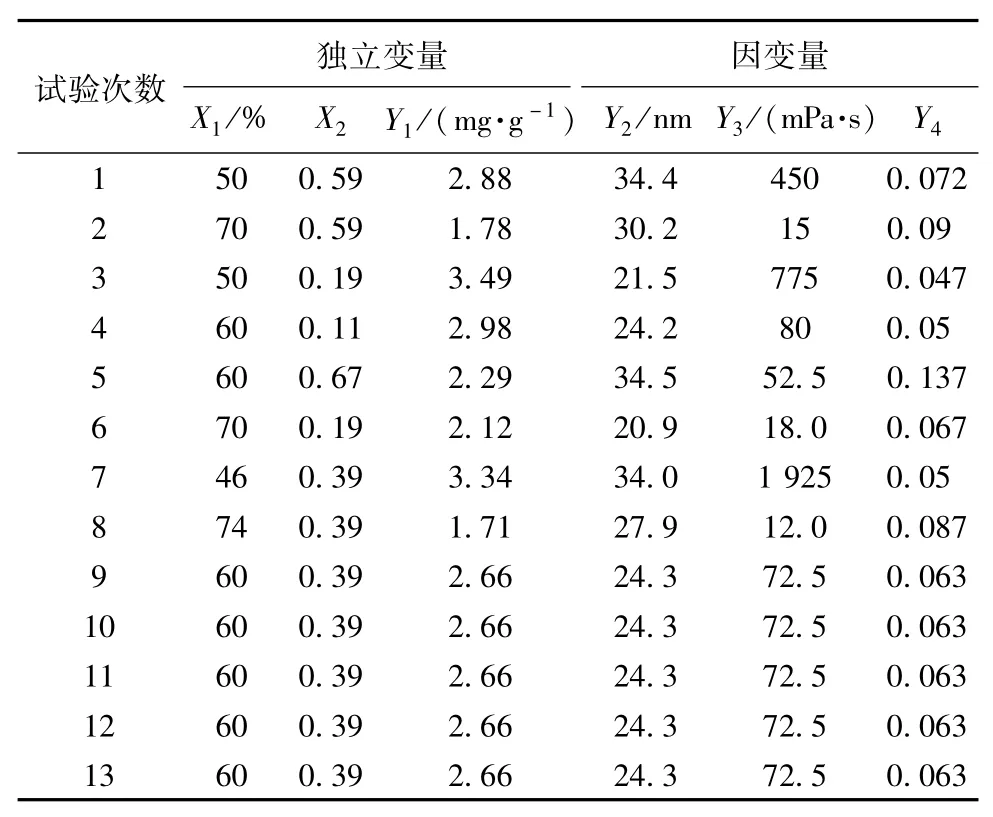
表2 试验中各因素的实际值以及各因变量结果
3.4 甘草次酸微乳的制备及含有量测定 根据优化处方,称取处方量的聚氧乙烯40氢化蓖麻油、油酸聚乙二醇甘油酯、二乙二醇单乙基醚,搅拌混合均匀后,加入甘草次酸药粉,待其溶解后慢慢滴加水相,匀速搅拌,得到澄清透明的甘草次酸含药微乳。平行制备3份,进行含有量测定,按“2.3”项下方法处理,测得微乳载药量为(3.00± 0.06)mg/g。
3.5 微乳性质考察 在微乳类型鉴别中发现,水溶性染料亚甲蓝的扩散速度比油溶性染料苏丹红快,表明本品为O/W型微乳。30℃恒温下的黏度为(0.089±0.003)Pa.s;28℃下pH值为(5.75±0.10);3批样品的平均粒径为(18.8±0.9)nm;多分散系数为(0.28±0.01),说明微乳粒径小,而且分布均匀,以15 000 r/min离心15 min,无分层现象,仍均一澄清。在相对湿度75%、温度25℃条件下,将该微乳放置0、1、3个月,发现仍保持澄清透明,未见油水分层现象。
4 讨论
伪三元相图是微乳制备的基础。本实验采用微孔酶标仪测定法绘制伪三元相图,临界点测定时可多个处方同时进行,临界突变点能够通过测定浊度值客观得到,比传统的目测滴定法更快速准确,实验效率显著提高。通过对优化处方进行验证发现,实测值与预测值基本相符,提示本实验处方优化方法是可行的。
药物的溶解度能够影响药物制剂的开发和药物的生物利用度。微乳作为一种新型的药物载体,能够提高难溶性药物的溶解度[15],它对药物的增溶作用取决于其组成成分混合表面活性剂、油相和水相对药物的溶解能力。在本实验中,甘草次酸为水难溶性成分,故混合表面活性剂和油相对于甘草次酸的溶解至关重要。因此,选择对甘草次酸溶解性比较好的聚氧乙烯40氢化蓖麻油、二乙二醇单乙基醚以及油酸聚乙二醇甘油酯分别作为微乳的表面活性剂和油相。然后,对甘草次酸微乳药物的含有量进行了测定,发现是其在水中溶解度(0.008 5 mg/g,自测值)的352倍,大幅提高了溶解性,提示微乳作为甘草次酸载体,有望显著改善其生物利用度,从而提高疗效。
参考文献:
[1] 金 敏,吴红金.甘草次酸药理作用的研究进展[J].医学综述,2009,15(11):1712-1715.
[2] 张明发,沈雅琴.甘草酸及其苷元甘草次酸的糖皮质激素样作用[J].现代药物与临床,2011,26(1):33-35.
[3] 张明发,沈雅琴,张艳霞.甘草及其有效成分的皮肤药理和临床应用[J].药物评价研究,2013,36(2):146-156.
[4] 郭波红,程 怡,林绿萍.甘草次酸平衡溶解度和表观油水分配系数的测定[J].广东药学院学报,2011,27(3):221-223.
[5] Monzer F.Microemu1sions:properties and app1ications[M]. Boca Raton:CRC Press,2008.
[6] 王 勤,李华文,彭新生.蛇床子素微乳的制备及其透皮能力的研究[J].中国药房,2010,21(27):2529-2531.
[7] 董文雪,何 军,杨亚妮,等.自微乳释药系统研究进展[J].中国医药工业杂志,2011,42(12):948-954.
[8] 郝淞瑶,杨燕云,张振秋,等.HPLC测定甘草提取物及苦芪甘片剂中甘草次酸的含量[J].辽宁中医杂志,2013,40 (11):2335-2337.
[9] LiW W,Yi S L,Wang Z H,et al.Se1f-nanoemu1sifying drug de1ivery system of persimmon 1eaf extract:Optimization and bioavai1abi1ity studies[J]. Int J Pharm,2011,420(1):161-171.
[10] Schmidts T,Nocker P,Lavi G,et al.Deve1opmentof an a1ternative,time and cost saving method of creating pseudoternary diagrams using the examp1e of a microemu1sion[J].Colloids Surf A,2009,340(1):187-192.
[11] 潘国梁,贾晓斌,魏惠华,等.药用微乳伪三元相图的几种制备方法比较研究[J].中国药房,2006,17(1):21-23.
[12] 王文娣,王 阳,王丽峰,等.星点设计-效应面法及正交实验设计优化淫羊藿提取工艺的比较研究[J].时珍国医国药,2010,21(11):2766-2768.
[13] 吴 伟,崔光华.星点设计-效应面优化法及其在药学中的应用[J].国外医学:药学分册,2000,27(5):292-298.
[14] 孙丹丹,徐新刚,生立嵩.星点设计-效应面法优化熊果酸自微乳给药系统[J].中国实验方剂学杂志,2013,19(8):46-50.
[15] Azeem A,Khan Z I,Aqi1M,etal.Microemu1sions asa surrogate carrier for derma1drug de1ivery[J].Drug Dev Ind Pharm,2008,35(5):525-547.
doi:10.3969/j.issn.1001-1528.2016.01.045
中图分类号:R944
文献标志码:B
文章编号:1001-1528(2016)01-0187-04
*通信作者:赵瑞芝(1968—),女,研究员,博士生导师,从事药物新剂型及其相关技术研究。Te1:13610241754,E-mai1:13610241754@163.comD
作者简介:肖志偲(1989—),女,硕士,从事药物新剂型及其相关技术研究。Te1:15989078630,E-mai1:xzc89152@163.com
基金项目:广东省自然科学基金团队项目(S2013030011515);广东省科技厅-广东省中医院联合专项项目(2011B032200009);广东省中医院项目(YK2013B1N11)
收稿日期:2014-09-01
猜你喜欢
现代畜牧科技(2021年8期)2021-10-13 07:21:42
中成药(2017年7期)2017-11-22 07:32:54
软件导刊(2016年9期)2016-11-07 18:25:50
中国科技博览(2016年22期)2016-11-01 15:45:01
商业会计(2016年15期)2016-10-21 08:43:39
职业(2016年10期)2016-10-20 21:38:59
天然产物研究与开发(2014年8期)2014-04-27 14:16:36
中成药(2014年9期)2014-02-28 22:28:49
