
闪式提取及高速逆流色谱联用提取高纯度甘草苷
2016-04-05 06:59:06徐向君余金鹏刘昌胜
中成药 2016年1期
徐向君, 余金鹏, 袁 媛, 刘昌胜
(华东理工大学教育部医用生物材料工程研究中心,上海200237)
闪式提取及高速逆流色谱联用提取高纯度甘草苷
徐向君, 余金鹏, 袁 媛, 刘昌胜*
(华东理工大学教育部医用生物材料工程研究中心,上海200237)
摘要:目的 研究从甘草渣中提取高纯度甘草苷的方法。方法 利用闪式提取法快速提取甘草黄酮类化合物,高速逆流色谱法分离出高纯度甘草苷。正交实验考察乙醇体积分数、固液比、提取时间、转速4个因素对提取率的影响。在最优工艺条件下制备出的甘草总黄酮经过初步纯化后,采用高速逆流色谱技术,以乙酸乙酯-甲醇-水(5∶3∶10)为两相溶剂体系,从中分离高纯度甘草苷。结果 闪式提取甘草渣中甘草总黄酮的优化条件为90%乙醇,料液比(g/mL)1∶40,提取时间3 min,转速16 000 r/min。从90 mg甘草总黄酮中分离得到35.71 mg甘草苷,纯度可达到94.7%,收率为87.8%。结论 建立了闪式提取和高速逆流色谱技术联用,用于制备高纯度甘草苷的方法。
关键词:甘草苷;闪式提取;高速逆流色谱
Isolation and purification of liquiritin from licorice residue by smashing tissue extraction and high-speed countercurrent chromatography
XU Xiang-jun, YU Jin-peng, YUAN yuan, LIU Chang-sheng*
(Engineering Research Center for Biomedical Materials,Ministry of Education,East China University of Science and Technology,Shanghai 2OO237,China)
KEY WORDS:1iquiritin;smashing tissue extraction;high-speed countercurrent chromatography(HSCCC)
1 前言
甘草(Glycyrrhizae Radix)是一种常用中草药,其中的活性成分主要有三萜类(以甘草酸为主),甘草黄酮类,甘草多糖类等[1]。近年来,随着甘草活性组分研究的深入,甘草黄酮类化合物也越来越受到人们的重视[2],特别是其中单组份化合物,很有希望被开发成新型小分子药物。如甘草苷是甘草中最主要的黄酮类物质,属于二氢黄酮类,具有很强的药理活性,对于抗抑郁有特殊的疗效[3],同时具有抗氧化作用、神经保护作用[4]、心肌细胞膜的保护作用[5]等。但甘草黄酮类化合物单组份的分离纯化,一直是甘草研究中的难点,这是因为甘草黄酮种类多、含有量低、易变性。
闪式提取(smashing tissue extraction)技术是一种通过高速剪切作用破坏植物组织,而快速提取其中有效成分的新型提取技术[6]。该技术能在室温条件、仅需几十秒到几分钟完成对有效成分的提取[7-8]。与传统提取技术相比,闪式提取技术更适用于黄酮、多酚等热敏性、易氧化有效成分的提取[9]。
高速逆流色谱(high-speed countercurrent chromatography,HSCCC)是一种新型液-液分配色谱技术,样品通过互不混溶的两相溶剂间分配系数的不同而在短时间内实现高效分离制备[10]。相对于传统的固-液柱层析技术,具有处理效率高、样品损失小、适用范围广等优点[11-12],被广泛地应用于制药、化工等各个领域。
本课题拟先用闪式提取技术提取甘草渣中的甘草总黄酮,再利用高速逆流色谱分离纯化出其中的甘草苷,以提高甘草的利用率和附加值。
2 材料与方法
2.1 原料与试剂 甘草来自宁夏拓明农业开发有限公司。甘草渣为提取甘草酸后的渣滓,经40℃真空干燥24 h以上制得。甲醇、乙酸乙酯、正丁醇、三氯甲烷、乙腈、醋酸、正己烷等均为分析纯,购自中国医药集团上海化学试剂公司。甘草苷对照品购自上海同田生物技术股份有限公司。
2.2 样品中甘草总黄酮的测定
2.2.1 标准曲线绘制 取甘草苷对照品0.022 0 g,精密称定,加入适量70%乙醇溶液超声溶解,定容至100 mL,混匀,配置成质量浓度为0.220 g/L的标准溶液。准确移取标准液0.1、0.2、0.4、0.6、0.8 mL于1~5号10 mL量瓶中,分别加入70%乙醇溶液1.9、1.8、1.6、1.4、1.2 mL,使总体积为2 mL,然后加入10% KOH溶液0.5 mL,混匀,室温放置15 min,待充分显色后,70%乙醇定容至10 mL,于410 nm波长处测定吸光度。以吸光度(A)为纵坐标,以甘草苷质量浓度(C)为横坐标,拟合得甘草总黄酮标准曲线A=28.6C+0.075 2,r=0.999 9。
2.2.2 样品中甘草总黄酮的测定[13]称取样品约0.02 g,70%乙醇溶液溶解,并定容至100 mL,量取1 mL样品溶液,置于10 mL量瓶中,依次加入70%乙醇溶液1 mL、10% KOH溶液0.5 mL,混匀,室温显色15 min,然后用70%乙醇定容至10 mL。于410 nm波长处测量吸光度,并根据标准曲线,计算样品溶液中总黄酮的质量浓度,进而计算甘草总黄酮的含有量。
2.3 样品中甘草苷的测定
2.3.1 色谱条件[14]Waters 2795高效液相色谱仪;使用Hypersi1C18色谱柱(250 mm×4.6 mm,5 μm,大连依利特分析仪器有限公司);流动相为乙腈-0.05%冰醋酸,梯度洗脱(0→10 min→15 min→20 min,20%→20%→38%→55%);检测波长250 nm;柱温20℃;体积流量1.0 mL/min。
2.3.2 标准曲线的绘制 准确称取5.5 mg甘草苷对照品,70%乙醇溶液溶解于25 mL量瓶中,配制成0.22 g/L的标准贮备液,取适量用流动相稀释成一系列质量浓度梯度(0.014、0.028、0.055、 0.11、0.22 mg/mL)的待测标准溶液,进样量为10 μL,记录色谱图,以峰面积(S)为纵坐标,甘草苷质量浓度(C,g/L)为横坐标进行线性回归,拟合得标准曲线为S =(4.50C-0.017 9)× 106,r=0.999 8。
2.3.3 样品中甘草苷的测定 称取样品约5 g,70%乙醇溶液溶解,并定容至25 mL,与标准曲线采用相同的色谱条件,根据峰面积计算样品中甘草苷含有量。
2.4 闪式提取甘草渣中甘草总黄酮 称取甘草渣0.5 g,按一定固液比加入乙醇水溶液,室温闪式提取几分钟,过滤,测定滤液中甘草总黄酮浓度,并计算提取率。正交试验设计法考察乙醇浓度(50%~90%)、固液比(1∶20~1∶40)、提取时间(1~3 min)、转速(10 000~16 000 r/min)等因素对提取率的影响。详见表1。

表1 闪式提取甘草总黄酮正交试验因素水平Tab.1 Factors and levels of orthogonal test for smashing tissue extraction of total glycyrrhiza flavonoids
2.5 甘草总黄酮提取物的初步纯化 将闪式提取所得提取液旋蒸干燥后,20倍体积去离子水加热溶解。将甘草总黄酮的水溶液于等体积乙醚中萃取3次,再于等体积乙酸乙酯中萃取3次,干燥除去溶剂后,得到甘草总黄酮初提物。初步纯化甘草总黄酮粗品后,HPLC法检测甘草苷。
选用AB-8树脂用于纯化甘草总黄酮,以2.12 mg/mL的上样质量浓度,pH为5,上样体积流量1.0 BV/h,以60%乙醇为洗脱剂,洗脱液经冷冻干燥挥去溶剂后,得到黄色粉末状甘草总黄酮粗提物500 mg,冷藏备用。
2.6 高速逆流色谱法分离纯化甘草苷
2.6.1 溶剂体系分配系数K的确定 一个稳定的两相溶剂体系是否适合于目标物质的分离,通常要看物质在该溶剂体系中的分配系数是否在一个合适的范围内[15]。根据高速逆流色谱分离黄酮类化合物的相关报道[16-17],选取了10种溶剂体系通过分配系数的测定来进行筛选。分配系数的测定方法:称取5.5 mg甘草黄酮样品,分别加入己达到分配平衡的上、下相5 mL,充分振荡溶解。用移液枪分别移取相同体积的溶有样品的上相溶液和下相溶液,待溶液挥干后,再用流动相溶解定容至3 mL。利用HPLC法,根据标准曲线测定溶剂体系上、下相所含样品的质量浓度,以上相中组分质量浓度CU与下相中组分质量浓度CL的比来求得样品在该溶剂系统中的分配系数K=CU/CL。
2.6.2 HSCCC分离方法 称取甘草总黄酮粗品90 mg,加入溶剂系统的上、下相各10mL,使之完全溶解,作为HSCCC的进样液。将上相以10 mL/min的速度泵入HSCCC分离管,待固定相充满整个分离管后,开启循环水浴,并将温度设定为25℃。再开启HSCCC主机,旋转方向为顺时针,使转速逐渐增加到800 r/min,同时以1.0 mL/min的流量泵入下相,待下相从柱出口流出,上相不再流出,表明上下相在分离管中达到平衡,由进样阀注人先前配制好的进样液,以250 nm波长进行监测,根据色谱图,手动收集各色谱峰组分。收集到的各组分样品溶液,蒸干后,用高效液相色谱法检测。
3 结果与讨论
3.1 闪式提取甘草渣中甘草总黄酮
3.1.1 正交试验结果 根据四因素三水平正交试验表,安排9组试验,每组重复3次以上,所得平均值列于表2,相应的误差线绘于图1。由图可知,所得数据的相对误差较小,重复性好,数据较为可靠。表2中,各因素同一水平的提取率求和得K1、K2、K3,相对应的平均值为k1、k2、k3,各因素的极差R为k1、k2、k3中最大值与最小值之差,可以反映该因素的影响大小。正交试验方差分析F的检验结果(表3)表明,4个因素对提取率的影响都不显著,可能是因为该实验存在一定的误差,且误差自由度较小,使检验的灵敏度低。因此,可从表2中的极差R中选择各因素对甘草总黄酮提取率的影响大小,依次为B(固液比)>A(乙醇浓度)>C(提取时间)>D(转速),而且B、A远大于C、D,这说明B(固液比)和A(乙醇体积系数)对提取率的影响要远大于C(提取时间)和D(转速)。由k1、k2、k3的大小可知,各因素的最优水平为A3B3C3D3,即闪式提取的最佳工艺条件为料液比(g/mL)1∶40,乙醇体积分数90%,提取时间3 min,转速16 000 r/min。闪式提取技术较传统提取方法具有省时、能室温提取、适用范围更广,不破坏成分、可选溶剂种类多,节能环保、操作简便等优点。

表2 闪式提取甘草总黄酮的正交试验Tab.2 Orthogonal tests for smashing tissue extraction of total glycyrrhiza flavonoids

图1 闪式提取甘草总黄酮的正交试验结果Fig.1 Orthogonal test results of smashing tissue extraction of glycyrrhiza flavonoids
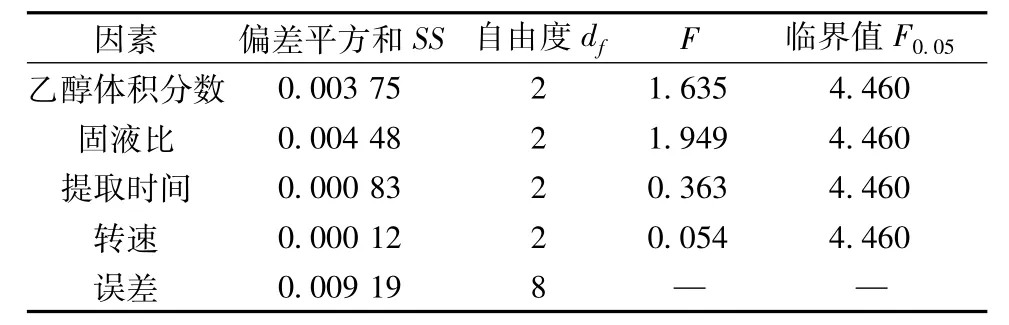
表3 方差分析Tab.3 Analysis of variance
3.1.2 验证试验 根据上述正交试验所得最佳工艺条件,进行3次验证试验,以A3B3C3D3为提取条件,得到甘草总黄酮提取率如表4所示。结果说明,正交试验所得工艺条件较好,且RSD为1.49%,表明该工艺稳定,重复性好。

表4 验证试验结果Tab.4 Verification test results
3.2 甘草总黄酮提取物的初步纯化 将闪式提取所得提取液旋蒸干燥后,20倍体积去离子水加热溶解。将甘草总黄酮的水溶液于等体积乙醚中萃取3次,再于等体积乙酸乙酯中萃取3次,干燥除去溶剂后,得到甘草总黄酮初提物。经过初步纯化的甘草总黄酮粗品使用HPLC检测,甘草苷含有量为169 mg/g。
选用AB-8树脂用于纯化甘草总黄酮,以2.12 mg/mL上样,pH为5,上样体积流量1.0 BV/h,以60%乙醇为洗脱剂,洗脱液经冷冻干燥挥去溶剂后,得到黄色粉末状甘草总黄酮粗提物500 mg,冷藏备用。甘草苷含有量为428 mg/g。
由于甘草黄酮各组分的极性相似,传统大孔树脂柱层析技术难以将其中的单一组分完全分离出来,因此需要更先进的技术(如高速逆流色谱法)进一步纯化其中的甘草苷。
3.3 高速逆流色谱法分离纯化甘草苷
3.3.1 溶剂体系的筛选 溶剂系统是决定HSCCC分离样品效果的最关键因素。不同的溶剂系统由于黏度、极性和密度等性质的差异,对相同的成分会产生不同的溶解、分配能力,因而形成分配系数的差异。根据色谱理论,利用HSCCC分离样品的必要条件是目标物能稳定的溶于两相溶剂;目标物在两相中具有合适的分配系数,必须在0.5和2之间。若远小于1,样品会很快随流动相流出,达不到分离效果;若远大于1,样品的出峰时间会延长,形成宽峰。根据表5的数据,应用乙酸乙酯-甲醇-水(5∶3∶10),乙酸乙酯-甲醇-水(3∶2∶5)、三氯甲烷-甲醇-n-丁醇-水(8∶8∶1∶4)作为两相时,K值在合适的范围内。其中,选取乙酸乙酯-甲醇-水(5∶3∶10)进行试验时,样品可以较好的从其他组分中分离出来,见表5。

表5 甘草苷在不同溶剂体系下的分配系数Tab.5 Partition coefficient of liquiritin in different solvent system s
3.3.2 HSCCC分离方法 使用乙酸乙酯-甲醇-水(5∶3∶10)作为两相溶剂系统。分离时间80 min,根据HSCCC图谱,第30分钟收集到组分,通过HPLC检测,组分A为目标物质(图2)。经干燥后,得到干燥物35.71 mg,收率为87.8%。获得甘草苷的纯度为94.7%(图3)。

A.甘草苷A.1iquiritin图2 HSCCC分离图谱Fig.2 Separation diagram of HSCCC

A.甘草苷A.1iquiriti图3 HPLC图谱Fig.3 HPLC chromatogram
对于成分较复杂的天然物质组分,传统的色谱技术难将其完全分离,高速逆流色谱技术具有高效分离、低费成本、工艺易放大、回收率高等优点,为天然药物复杂混合物中特定活性成分的高纯度单体的制备与分离纯化带来了更优的解决方案。
4 结论
本实验采用闪式提取与高速逆流色谱联用的方法来制备高纯度甘草苷。闪式提取正交优化的最佳工艺条件为乙醇体积分数90%,料液比(g/mL)1∶40,提取时间3 min,转速16 000 r/min。经初步纯化后,以乙酸乙酯-甲醇-水(5∶3∶10)为HSCCC两相溶剂系统,进一步分离纯化甘草苷,一次进样甘草总黄酮初提物90 mg,可得到35.71 mg甘草苷,纯度为94.7%。因此,所建立的闪式提取及HSCCC联用的方法用于分离纯化甘草渣中的甘草苷是可行的,该方法在快速提取、分离和纯化天然产物方面具有较好的发展潜力和广泛的应用前景。
参考文献:
[1] 马海琴,王志清.甘草的开发与利用[J].特种经济动植物,2005,8(10):22-23.
[2] 季宇彬,姜 薇,范玉玲,等.甘草黄酮的研究进展[J].中草药,2004,35(9):附录5-附录6.
[3] 肖 渊.甘草苷的抗抑郁作用及其机理研究[D].北京:北京中医药大学,2009.
[4] 杨 云,卞广兴,吕秋军.甘草苷对原代海马神经细胞的保护和营养作用[J].中国中药杂志,2008,33(8):931-935.
[5] 董 晞,赵世萍,刘 岩,等.甘草苷对乌头碱致心肌细胞损伤的保护作用[J].中华中医药杂志,2009,24(2):163-166.
[6] 刘延泽.植物组织破碎提取法及闪式提取器的创制与实践[J],中国天然药物,2007,5(6):401-407.
[7] 邓引梅,宋发军,崔永明,等.甘草叶总黄酮提取工艺[J].中南民族大学学报:自然科学版,2008,27(1):41-43.
[8] 邓引梅,崔永明,李 唯,等.响应面法优化闪式提取甘草叶总黄酮工艺研究[J].化学与生物工程,2008,25 (9):44-47.
[9] 孟庆举,刘晓谦,杨 华,等.闪式提取技术的进展[J].中国实验方剂学杂志,2013,19(19):349-354.
[10] 周婷婷,范国荣.高速逆流色谱在天然产物活性成份分离制备中的应用[J].中国药科大学学报,2007,38(5):391-395.
[11] Jiang Y,Lu H T,Chen F.Preparative purification of g1ycyrrhizin extracted from the root of 1iquorice using high-speed counter-current chromatography[J].J Chromatogr A,2004,1033(1):183-186.
[12] 彭会咏,范国荣,吴 玉.白花败酱草黄酮类成份的高速逆流色谱快速制备[J].中国药学杂志,2006,41(13):977-979.
[13] 马 玲,安 瑜,王 坤,等.甘草中总黄酮含量测定的方法学研究[J].宁夏医学杂志,2010,32(5):428-430.
[14] 黄辉球,刘 强,江 洁.一种甘草及其制剂的质量控制方法:中国,CN201310037821.8[P].2013-06-26.
[15] Wu H,Feng R H,Guan S G,et al.Rapid preparative iso1ation of a new pheny1propanoid g1ycoside and four minor compounds from Sparganium stoloniferum using high-speed countercurrent chromatography as a fractionation too1[J].J Sep Sci,2012,35(9):1160-1166.
[16] Xiao X H,Si X X,Tong X,et al.Preparation of f1avonoids and diary1heptanoid from Alpinia katsumadai hayata by microwave-assisted extraction and high-speed counter-current chromatography[J].Sep Purif Technol,2011,81(3):265-269.
[17] 胡昌盛,彭金咏.采用高速逆流色谱法快速制备枳壳中高纯度柚皮苷和新橙皮苷的研究[J].临床合理用药杂志,2013,33(6):58-60.
*通信作者:刘昌胜(1967—),男,教授,从事生物材料研究。Te1:13801823622,E-mai1:1iucs@ecust.edu.cn
作者简介:徐向君(1990—),男,硕士,从事中草药的提取纯化工作。Te1:15026535586,E-mai1:xxj0768@163.com
收稿日期:2015-03-23
doi:10.3969/j.issn.1001-1528.2016.01.015
中图分类号:R284.1
文献标志码:A
文章编号:1001-1528(2016)01-0072-05
