
甲醛合成乙醇醛研究及其应用进展
2016-03-20 05:09:49李青松于记生
天然气化工—C1化学与化工 2016年5期
辛 坤,李青松,贾 冰,于记生
(中国石油大学(华东)重质油国家重点实验室 山东 青岛 266580)
甲醛合成乙醇醛研究及其应用进展
辛 坤,李青松,贾 冰,于记生
(中国石油大学(华东)重质油国家重点实验室 山东 青岛 266580)
介绍了乙醇醛作为中间体在食品、医药卫生和化工等行业的应用。综述了以甲醛为原料生产乙醇醛的工艺,包括氢甲酰化反应;高选择性的formose反应;通过典型的formose反应制备糖醇,再含水热解;N-杂环卡宾催化。指出N-杂环卡宾催化甲醛合成乙醇醛是最具前景的生产策略,值得深入研究。
乙醇醛;甲醛;氢甲酰化;formose反应;N-杂环卡宾
乙醇醛(HOCH2CHO)又名羟基乙醛,是最小的糖分子。其与丙烯醛发生反应后能形成核糖,而这正是RNA的重要成分,它是生命起源过程中非常关键的糖分子[1]。其分子中有醛基和羟基两种官能团,具有醇和醛的双重性质,化学性质活泼。作为重要的化学中间体,广泛应用于食品、医药卫生、化工等行业。以甲醛为原料生产乙醇醛,意义重大,且尚未学者进行详细评述。
1 应用现状
乙醇醛的下游产品多是市场紧俏并具有高附加值的精细化工产品。同时,若可降低乙醇醛生产成本,则下游产品范围及产量将进一步扩大。
1.1 食品行业的应用
蛋白质、氨基酸等氨基化合物和醛、酮等与还原糖相遇,经过一系列反应生成褐色聚合物。国外对褐化反应应用于食品香精生产之中进行了广泛研究,在肉类香精及烟草香精中取得了很好的效果。而乙醇醛作褐化试剂效果显著[2],是优良的食物染色、调味品、香精等的褐变启动子。
硼氢化钠作用下,乙醇醛很容易和氨基反应,可选择性修饰蛋白质的氨基,是一种优异的修饰剂[3]。通过还原烷基化,乙醇醛还可作为蛋白材料或其它含氨基材料的交联介质,如可应用于强化香肠肠衣等。
1.2 医药卫生行业的应用
对乳腺癌细胞的研究表明[4],乙醇醛可以抑制葡萄糖-6-磷酸脱氢酶、甘油醛-3-磷酸脱氢酶和铜、锌超氧化物歧化酶的活性,并抑制细胞生长,最终诱导其凋亡,乙醇醛可能是中性粒细胞抗肿瘤活性的重要介质。
1.3 化学工业的应用
生物质热解得到的生物油中存在大量的乙醇醛。生物油中的乙醇醛转化为1,1,2-三甲氧基乙烷[5],其辛烷值较高且沸点在汽油沸程内,与适量C1~C4醇混合可提高汽油辛烷值;Yeh等[6]研究表明1,1,2-三甲氧基乙烷是一种理想的柴油添加剂;Surampudi等[7]研究结果发现,1,1,2-三甲氧基乙烷无毒、腐蚀性小、饱和蒸汽压低、氧化速率快,是一种优异的燃料电池燃料。因此,由乙醇醛生产高附加值的1,1,2-三甲氧基乙烷是一种很好的选择。
乙醇醛加氢可制备大宗化学品乙二醇。乙二醇是一种重要的化工原料,主要用于生产聚酯纤维、防冻剂、炸药、涂料、特种溶剂等,用途十分广泛。若可实现乙醇醛的廉价工业生产,则此工艺前景广阔。
1.4 生产现状
目前市场上出售的乙醇醛多以二聚体形式存在,价格昂贵。国外乙醇醛已得到广泛研究与应用,而国内相关报道很少。乙醇醛的生产还处在实验室阶段,为实现大规模工业生产,找到一种简单廉价的生产工艺至关重要。
2 乙醇醛的生产工艺
随着C1化学的发展与卡宾催化剂的不断深入研究,国外乙醇醛的合成有新的进展,从而使乙醛醛生产有着更加广阔的市场前景。以甲醛为原料,生产乙醇醛工艺可分为:氢甲酰化法,选择性formose反应,经formose反应生成的糖醇含水热解及N-杂环卡宾催化。
2.1 甲醛氢甲酰化
1938年,德国化学家Roelon O等发现高温高压、钴催化剂作用下,烯烃与CO和H2反应可以生成醛(如图1(a)所示)。该过程称为氢甲酰化或醛化反应,系指烯烃双键加了一个醛(H-CHO)。目前,烯烃的氢甲酰化工业上已广泛应用,铑或钴等的络合物是该反应有效的催化剂[8]。当使用甲醛的C=O键代替烯烃的C=C键作反应基质时,发生甲醛的氢甲酰化反应(如图1(b)所示)。该反应[9]首先涉及到CO插入金属-碳键,随后酰基金属络合物在含活性H的亲核试剂(HY)作用下发生氢解、水解、醇解或相关反应,如图2所示。

图1 (a)CO和H2反应(b)甲醛的氢甲酰化反应Fig.1 (a)Reaction of CO with H2(b)Hydroformylation of formaldehyde

图2 甲醛氢甲酰化机理示意图[9]Fig.2 Proposed catalytic cycle of the hydroformylation of formaldehyde[9]
由于Co活性高且不易中毒失活,初期氢甲酰化多用含Co催化剂,如金属钴、钴的氧化物、钴盐等。反应宜在110~180℃、1.0~49.0MPa下进行,以氮化物、卤化物、氧化物或磷化物作促进剂。Yukawa等[10]以Co2(CO)8为催化剂,n(H2)/n(CO)=1,二氧六环作溶剂,并加入一定量DMF,在120℃,19.7MPa下反应20min,乙醇醛收率最高可达50%。
但Co体系反应条件苛刻,投资成本高,人们意识到基于铑氢甲酰化催化剂的潜在优点超过了含钴的催化剂,可有效地在更温和的温度和压力下操作,1980年10%的氢甲酰化反应以铑作催化剂,1995年已经增长到80%,Co体系逐渐被工业淘汰。以铑的配位化合物做催化剂[18],如三烷基亚磷酸盐、三芳基亚磷酸盐或三芳基磷配体,与CO复合物作催化剂,是甲醛氢甲酰化高效的催化剂。过量的磷配体可稳定催化剂,虽会降低其活性,但加入少量的胺即可弥补。
孟山都公司[12]以铑的羰基、取代磷及氯络合物作为催化剂,大大提高了聚甲醛甲酰化生成乙醇醛的反应速率。以RhCl(CO)(PPh3)2作催化剂,在110℃,28.5MPa,n(H2)/n(CO)=1,四甘醇二甲醚作溶剂,加入少量三乙胺的条件下,乙醇醛收率达73%,选择性为78%。通过研究溶剂对甲酰化反应的影响[13],发现乙腈是较好的溶剂,合适的条件下,甲醛转化率和乙醇醛的收率分别可达98%与78%。若增大n(H2)/n(CO)=2,将进行深度加氢获得高收率的乙二醇。
考虑到Co盐可溶于有机溶剂中,而Rh的磷化物在有机溶剂中溶解性很差,使用基于Co-Rh体系的催化剂可以阻止Rh因原料中的硫等毒物而失活,但活性较差。Marchionna等[14]使用羰基钴和羰基铑作催化剂,DMF作溶剂,n(H2)/n(CO)=1,110℃、13.4MPa下反应,但乙醇醛收率仅为26%。
甲醛的氢甲酰化反应,原料廉价易得。其中基于铑的催化剂反应条件缓和,乙醇醛收率高,优势明显。但铑的价格昂贵,目前而言,发展铑回收技术将催化剂的损失限制到最小程度或找到廉价高活性的催化剂,是今后的研究方向。
2.2 选择性的formose反应
自1861年Butlerow首次报道甲醛聚糖反应(formose反应)以来,尽管人们不断探索提高目的产物的选择性,但研究进展缓慢。Weiss等[15]以Ca(OH)2为催化剂催化甲醛水溶液聚合,发现产物中主要是C2~C7的直链糖和支链糖(粘稠糖浆),组分多达47种。其中,约50%为C6糖,约25%为C5糖,约10%为C7糖,同时存在甲酸盐、甲醇等副产物,无法通过普通方法对乙醇醛进行分离提纯,无工业应用价值。
经formose反应直接选择性制备乙醇醛的报导较少,Weiss等[16]以甲醛水溶液为原料,催化反应在装填分子筛或沸石的喷淋床中进行。质量分数30%的甲醛水溶液与等体积的NaOH水溶液缓慢的通过 NaX、5A或 Na沸石床层,操作条件 94℃、0.1MPa、液体体积空速(LHSV)1.21~2.36h-1,通过将pH保持在11左右以维持分子筛稳定。体系中主要反应如图3所示,利用分子筛的择型性,采取合适的反应条件,反应选择性可以严格控制,副产的其它糖可控制在1%以下,乙醇醛收率可达75%。但需严格控制工艺条件,存在设备腐蚀严重,产生大量废水等问题,应用于工业生产还需进一步探究。

图3 Weisis[16]选择性制备乙醇醛相关反应Fig.3 The reactions in preparation of glycolaldehyde put forward by Weisis[16]
2.3 糖醇的含水热解
乙醇醛是木质纤维素材料单糖或多糖的热解产物,Majerski等[17]将w(H2O)为30~60%的糖溶液喷入520~560℃的反应器中,停留时间0.5~2s,以单分子醛糖如葡萄糖为原料,乙醇醛收率可达50%。水进攻醛糖羟基上的氢,经过电子转移与化学键的断裂得到乙醇醛,机理如图4所示。
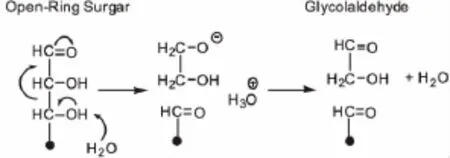
图4 醛糖的含水热解[17]Fig.4 Hypothetical water-catalyzed retro-aldo scission of an aldose[17]
以甲醛为原料,通过两步可以得到较高收率的乙醇醛。首先,甲醛在无机碱作用下反应得到糖醇,其为醛糖和酮糖的混合物,碱性条件下二者相互转化[18],控制条件可选择性的得到醛糖(如图5所示)。Weiss等[19]通过控制甲醛转化率,生成物中葡萄糖选择性可达75%;然后,将得到的糖浆经减压蒸馏分离后与适量水混合,喷入高温的反应器中热解即可得到一定收率的乙醇醛。

图5 单糖在碱性介质中的异构化[18]Fig.5 Isomerization ofmonosaccharides inalkaline medium[18]
此工艺催化剂无机碱廉价易得,且选择性的得到醛糖反应条件较易控制,但含水热解需高温能耗较高,同时存在设备腐蚀和废水处理问题,限制了该工艺的发展。
2.4 N-杂环卡宾催化
N-杂环卡宾的研究历史最早要追溯到1943年,Ukai等[20]发现维生素B1也可催化苯甲醛的安息香缩合反应,其操作简单,污染小,无毒无害,成为了氰化物的最佳替代试剂。迄今为止,经典的氮杂环卡宾(NHC)共有四类:(A)噻唑类;(B)三唑类;(C)咪唑类;(D)咪唑啉类(如图6所示)。

图6 N-杂环卡宾结构Fig.6 Structure of NHC
1980年以来,以Matsumoto[21]为代表的科学家们针对早期甲醛聚糖反应存在的缺陷,对甲醛聚糖反应进行了深入的研究,取得了令人瞩目的进展。以噻唑盐为催化剂,三碳糖的选择性已从早期的10%以下提高到接近90%,并进行了一系列技术上的重大改进。要选择性的生成乙醇醛,则需其它类型的NHC催化剂。
2.4.1 三氮唑卡宾催化
Teles等[22]将由噻唑盐、咪唑盐和三氮唑盐得到的稳定的卡宾催化甲醛反应,发现基于1,4-二取代-1H-1,2,4-三氮唑盐的催化剂乙醇醛为主要产品。Teles分离出三氮唑催化甲醛安息香缩合循环中的几个中间体,提出反应机理如图7所示:三氮唑盐1中具有催化活性2位碳原子上的氢原子由于受到相邻氮原子的影响,具有很强的酸性,在弱碱的作用下很容易被去质子化,产生的碳负离子以更稳定的氮杂环卡宾存在。第一分子的甲醛在碳负离子的催化下,羰基碳原子由传统的正电性转变为负电性(即醛基的反应极性发生了翻转),进而对第二分子的甲醛进行亲核加成,最终得到产物乙醇醛。

图7 三氮唑盐催化甲醛缩合机理[22]Fig. 7 Mechanism of formaldehyde condensation catalyzed by triazolium salts[22]
Grubbs[23]等研究发现三氮唑盐的甲醇加成产物制备的卡宾催化剂具有很高的活性,同时对水和氧有一定的忍耐性。减压下加热处理4,5-二氢-5-甲氧基-1,3,4-三苯基-1-氢-1,2,4-三氮唑,即得到高活性的三氮唑卡宾1,3,4-三苯基-4,5-二氢-1H-1,2,4-三氮唑-5-亚基。此卡宾催化剂在无水隔绝空气的环境中,150℃以下可长期稳定存在,可与醇、氧气等反应[24],是第一个可商品化的卡宾催化剂[25],为三氮唑卡宾的工业化应用奠定了坚实的基础。将其用于甲醛安息香缩合反应,催化剂用量为物质的量分数0.5%, DMF为溶剂,80℃下反应20min,乙醇醛收率可达60%,选择性达85%(图8),延长反应时间会造成乙醇醛的深度转化导致其收率降低。

图8 1,3,4-三苯基-4,5-二氢-1H-1,2,4-三氮唑-5-亚基的制备及用于催化甲醛缩合[23]Fig.8 Preparation of1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene and its catalysis in formaldehyde condensation[23]
当1,4-二取代-4H-1,2,4-三氮唑卡宾中4号位被小分子取代基取代,其催化甲醛缩合同样可获得较高的乙醇醛收率。US 5508422[26]中以2-对硝基苯基-1-甲基-1,2,4-三唑碘化物(如图9所示)作催化剂催化甲醛反应,四氢呋喃为溶剂,与催化剂等物质量的三乙胺作助碱,氮气保护下80℃下反应2h,产物乙醇醛收率46.2%,副产物DHA和甘油醛收率分别为0.7%和4.1%。但可以看出需要较长的反应时间,催化剂活性较差。
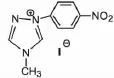
图9 2-对硝基苯基-1-甲基-1,2,4-三唑碘化物结构Fig. 9 Structure of 1-p-Nitrophenyl-4-methyl-1,2,4-triazolium iodide
常规的三氮唑卡宾催化甲醛缩合,活性高且选择性好,被广泛研究,但合成步骤繁琐[27]、生产成本较高,制备工艺有待进一步开发优化。Gehrer等[28]研究了Ⅰ结构(如图式10所示)催化剂对甲醛缩合反应的催化效果,研究表明Ⅱ(Nitron)可催化甲醛选择性生成乙醇醛和甘油醛且活性高,当甲醛/Niron物质的量比大于400时产物中乙醇醛选择性达90%以上。以DMF为溶剂,加入等物质量的Nitron和助碱三乙胺,氮气保护下80℃搅拌1h,乙醇醛收率可达65.2%,甘油醛收率16.0%,DHA收率0.8%,C4收率1.2%。当使用原位生成的Nitron作催化剂时[29](如图10所示),反应活性很高且无需添加助碱,100℃反应1h,乙醇醛收率45.2%,甘油醛收率42.4%,DHA收率0.7%,C4收率6.2%。Nitron与1,3,4-三苯基-1,2,4-三氮唑盐相比具有相似的活性且合成工艺简单,但存在乙醇醛选择性相对较差的问题。找到耐水耐氧的高活性的三氮唑卡宾,并优化其生产工艺,是广大化学家的共同目标。

图10 Nitron催化甲醛缩合[28,29]Fig.10 Formaldehyde condensation catalyzed by Nitron[28,29]
2.4.2 咪唑卡宾催化
均相的咪唑盐催化甲醛缩合主要产物为DHA且收率不高,几乎没有乙醇醛产生。低的甲醛转化率归咎于咪唑卡宾催化剂在反应中耦合,反应活性降低(如图11所示)。

图11 咪唑卡宾耦合失活Fig.11 Inactivation caused by coupling of imdazolium carbene

图12 1,3-二(2,6-异丙基苯基)氯化咪唑催化甲醛缩合Fig.12 Formaldehyde condensation catalyzed by 1,3-bis (2,6-di-isopropylphenyl)imidazolium chloride
Vetter等[30]通过控制催化剂活性位和优化反应条件成功使用咪唑盐催化制备出乙醇醛。加入一定量的多聚甲醛、四氢呋喃、三乙胺和 1,3-二(2,6-异丙基苯基)氯化咪唑,在带搅拌的三口烧瓶中加热至80℃反应1h,甲醛转化率为30%,乙醇醛选择性为89%(如图12所示)。
由于咪唑杂环化合物C2位酸性较弱,即使控制催化剂活性位其催化活性还是较低。但一些报导描述了高活性的负载咪唑的催化剂具有合成利用卡宾催化剂的稳定性增加,并超过其他杂环系统[31]。Storey等[32]使用[(4-氯甲基)-5-苯基]戊基苯乙烯和N-甲基咪唑结合制备Ⅰ,负载量0.89mmol/g;使用氯化王树脂与N-甲基咪唑结合制备Ⅱ,负载量1.32mmol/g(如图13所示)。反应在带搅拌的两口烧瓶中进行,以四氢呋喃为溶剂,NaOH水溶液为助碱,加热至回流。当使用Ⅰ作催化剂,反应10min乙醇醛收率可达77%,当使用Ⅱ作催化剂,反应15min乙醇醛收率可达72%。Ⅰ作催化剂循环使用时,每一循环产物收率下降10%左右,反应时间可由数小时、数天到数分钟。
咪唑卡宾通过引入合适的取代基,特别是可固载在树脂等载体上[33],具有较高的活性及选择性,同时固载的咪唑盐有利于实现产品分离与催化剂的循环使用,有很好的应用前景。

图13 固载的咪唑催化剂[32]Fig.13 Solid-supported imidazolium catalysts[32]
3 总结与展望
四种制备策略各有优缺点,基于铑催化的甲醛氢甲酰化反应虽反应条件缓和但仍需较高的压力,且铑价格昂贵;选择性的formose反应与糖醇的含水热解虽然催化剂无机碱廉价易得,但需严格控制反应条件,且会产生大量废水,发展必须解决废水的处理问题。相比而言N-杂环卡宾催化甲醛自缩合,其催化效率高、选择性好、产物易分离且稳定性较好,最具开发应用前景。然而,由于N-杂环卡宾的发展和研究起步较晚,目前对其催化机理的研究还不深入系统,具有较好实际应用价值的研究还不多,急需发展催化性能更为优异的新型NHC催化剂的合成及应用研究,这对实现乙醇醛工业化有重要意义。
[1]Ricardo A,Carrigan M A,O lcott A N,et al.Borate minerals stabilize ribose[J].Science,2004,303:196-196.
[2]Ruiter A.Color of smoked foods[J].Food Technol,1979, 33:54-58.
[3]Geoghegan K F,Ybarra D M,Feeney R E.Reversible reductive alkylation of amino groups in proteins[J]. Biochemistry,1979,18:5392-5399.
[4]Al-Maghrebi M A,Al-Mulla F,Benov L T.Glycolaldehyde induces apoptosis in a human breast cancer cell line[J]. Arch Biochem Biophys,2003,417:123-127.
[5]Li W,Pan C,Zhang Q,et al.Upgrading of low-boiling fraction of bio-oil in supercritical methanol and reaction network[J].Bioresour Technol,2011,102:4884-4889.
[6]Yeh I C,Schlosberg R H,Miller R C,et al.Diesel fuel composition[P].US:6447557,2002.
[7]Frank H A,Halpert G,Narayanan S R,et al.Aqueous liquid feed organic fuelcellusing solid polymer electrolyte membrane[P].US:5599638A,1997.
[8]Franke R,Selent D,Borner A.Applied hydroformylation [J].Chem Rev,2012,112:5675-5732.
[9]Chan A S C.A Mechanistic Approach to the study of homogeneous catalytic hydroformylation of formaldehyde [J].Comment Inorg Chem,1993,15:49-65.
[10]Yukawa T,Wakamatsu H.Method of producing glycolaldehyde[P].US:3920753,1975.
[11]Chan A S C,Carroll W E,Willis D E.Rhodiumcatalyzed hydroformylation of formaldehyde[J].J Mol Catal,1983, 19:377-391.
[12]Chan A S C.Hydroformylation process to prepare glycolaldehydes[P].US:4477685,1984.
[13]Goetz R W.Process for preparing glycolaldehyde and/or ethylene glycol[P].US:4405821,1983.
[14]Marchionna M,Longoni G.Hydroformylation of formaldehyde catalyzed by cobalt-rhodium bimetallic systems[J].J Mol Catal,1986,35:107-118.
[15]Weiss A H,Lapierre R B,Shapira J.Homogeneously catalyzed formaldehyde condensation to carbohydrates[J]. J Catal,1970,16:332-347.
[16]Weiss A H,Trigerman S,Dunnells G,et al.Ethylene glycol from formaldehyde[J].Ind Eng Chem Process Des Dev,1979,18:522-526.
[17]Majerski P A,Piskorz J K,Radlein St.D A G. Production of glycolaldehyde by hydrous thermolysis of sugars[P].US:7094932 B2,2006.
[18]Angyal S J.The Lobry de Bruyn-Alberda van Ekenstein transformation and related reactions[J].Top Curr Chem, 2001,215:1-14.
[19]Weiss A H,Socha R F,Likholobov V A,et al.Formose sugars from formaldehyde[J].Appl Catal,A,1981,1: 237-246.
[20]Ugai T,Tanaka S.S Dokawa.A new catalyst for acyloin condensation[J].Yakugaku Zasshi,1943,63:269-300.
[21]Matsumoto T,Yamamoto H,Inoue S.Selective formation of triose from formaldehyde catalyzed by thiazolium salt [J].JACS,1984,106:4829-4832.
[22]Teles J H,Melder J P,Ebel K,et al.The chemistry of stable carbenes.Part 2.benzoin-type condensations of formaldehyde catalyzed by stable carbenes[J].Helv Chim Acta,1996,79:61-83.
[23]Grubbs R H,Romero P E.A combined formose/transfer hydrogenation process for ethylene glycol synthesis[P]. WO:2009049114 A1,2009.
[24]Enders D,Breuer K,Raabe G,et al.Preparation,structure, and reactivity of 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene,a new stable carbene[J].Angew Chem Int Ed,1995,34:1021-1023.
[25]Enders D,Balensiefer T.Nucleophilic carbenes in asymmetric organocatalysis[J].ChemInform,2004,35:534-41.
[26]Teles J H,Melder J P,Gehrer E,et al.Addition products of triazolium salts[P].US:5508422,1996.
[27]Enders D,Breuer K,Kallfass U,et al.Preparation and application of 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene,a stable carbene[J].Synthesis,2003,34: 1292-1295.
[28]Gehrer E,Harder W,Ebel K,et al.Catalytic preparation of formaldehyde condensates[P].US:5298668,1994.
[29]Kriven’ko A P,Morozova N A.Synthesis of 1,4-diphenyl-3-phenylimino-1,2-dihydro-1,2,4-triazolium hydroxide (Nitron)[J].Russ J Appl Chem,2006,79:506-507.
[30]Vetter A J,Janka M E,Zoeller J R.Coupling of formaldehyde to glycoaldehyde using N-heterocyclic carbene catalysts[P].US:7498469B1,2009.
[31]Gao G,Xiao R,Yuan Y,et al.Efficient imidazolium catalysts for the benzoin condensation[J].J.Chem Res, 2002,33:27-27.
[32]Storey J M D,Williamson C.Imidazole based solidsupported catalysts for the benzoin condensation[J]. Tetrahedron Lett,2005,46:7337-7339.
[33]Kim D W,Chi D Y.Polymer-supported ionic liquids: imidazolium salts as catalysts for nucleophilic substitution reactions including fluorinations[J].Angew Chem Int Ed,2004,43:483-485.
Progress in preparation of glycolaldehyde from formaldehyde and its application
XIN Kun,LI Qing-song,JIA Bing,YU Ji-sheng
(State Key Laboratory of Heavy Oil Processing,China University of Petroleum(East China),Qingdao 266580,China)
Glycolaldehyde is an intermediate to produce high value-added products and can be used in food,medicine,hygiene and chemical industry.The paper summarizes the synthesis of glycolaldehyde,including hydroformylation of formaldehyde,selective formose reaction,hydrous thermolysis of sugar alcohols produced by typical formose reaction and N-heterocyclic carbene catalysis. Each process is reviewed.It is pointed out that N-heterocyclic carbene catalysis is the most promising production strategy,which is worthy of further research.
glycolaldehyde;formaldehyde;hydroformylation;formose reaction;N-heterocyclic carbene
TQ224.6
:A
:1001-9219(2016)05-88-07
2015-12-01;
:国家自然科学基金项目(No.51172284),中央高校基本科研业务费专项资金(No. 15CX06048A)资助项目;
:辛坤(1991-),男,硕士研究生;*联系人:李青松,博导,从事化工分离工程、石油天然气加工、化工材料和新能源与煤化工等领域的研究,电话0532-86981853,E-mail:licup01@163.com。
猜你喜欢
中国饲料(2021年17期)2021-11-02 08:15:14
科学技术与工程(2020年34期)2021-01-08 05:43:32
世界农药(2019年4期)2019-12-30 06:25:08
广东饲料(2016年5期)2016-12-01 03:43:22
合成化学(2015年2期)2016-01-17 09:03:25
合成化学(2015年9期)2016-01-17 08:57:21
合成化学(2015年10期)2016-01-17 08:56:37
时尚北京(2015年1期)2015-01-30 00:00:35
应用化工(2014年1期)2014-08-16 13:34:08
无机化学学报(2014年6期)2014-02-28 17:31:59
