
β-环糊精液相色谱固定相拆分和测定梨中烯唑醇对映体含量
2016-02-23 07:37:26曹志刚李来生程彪平张宏福
分析测试学报 2016年1期
曹志刚,李来生,2*,程彪平,张宏福,曾 春,谯 婷
(1.南昌大学 化学学院,江西 南昌 330031;2.南昌大学 分析测试中心,江西 南昌 330047)
β-环糊精液相色谱固定相拆分和测定梨中烯唑醇对映体含量
曹志刚1,李来生1,2*,程彪平1,张宏福1,曾春1,谯婷1
(1.南昌大学化学学院,江西南昌330031;2.南昌大学分析测试中心,江西南昌330047)
摘要:制备了一种单脲基衍生化β-环糊精键合有序介孔SBA-15手性固定相(UCDP),并将其填充于色谱柱中,以甲醇和水为流动相,成功地拆分了手性农药烯唑醇对映体。考察了流动相组成和温度等因素对手性分离的影响,确定最佳条件为:流动相为甲醇-水(40∶60),流速0.5 mL/min,柱温25 ℃,检测波长248 nm。在优化分离的基础上,建立了一种高效液相色谱快速测定梨中烯唑醇对映体残留量的新方法。梨样品经乙腈提取,氨基固相萃取柱净化后按上述条件测定。单个烯唑醇对映异构体在0.5~125 μg/mL范围内呈良好的线性关系。梨样品中分别添加1.25 μg/mL和5.0 μg/mL两水平的烯唑醇对映体,测得R-烯唑醇的平均回收率分别为92.0%和94.4%,S-烯唑醇的平均回收率分别为91.2%和92.8%。按3倍于噪音信号推算得到单个烯唑醇的检出限为0.02 μg/mL。该环糊精类固定相的自制方法简便,脲键固定相更稳定,制备成本较低。所建立的对映体分析方法快速、准确、重现性好,在梨等果蔬样品中农药对映体残留量检测方面有良好的应用前景。
关键词:高效液相色谱法;环糊精手性固定相;烯唑醇对映体;手性农药;食品安全
分子中含有不对称的原子、不对称中心、不对称面的化合物均可能具有手性(Chirality),手性是自然界物质的基本属性之一。除对偏振光作用后结果不同外,对映体的其他物理性质和化学性质几乎相同,但在与手性的生命系统作用时常表现出明显的差异[1-2]。近年来,手性农药对映异构体的杀虫活性、生物毒性、体内代谢的差异性越来越受到国际社会的高度重视。例如:除草剂异丙甲草胺,其S-型对映体有很好的除草活性,而R-型对映体不仅无除草活性,还有致癌作用[3]。常见的三唑类手性农药对映体不仅杀菌活性不同,对大鼠的肝脏毒性也不同,已被美国环境保护署(EPA)列为潜在的手性致癌物[4-5]。因此,研发新型的手性固定相,并建立测定手性农药对映体含量的新方法,对深入了解对映体的杀菌活性和人体毒性,乃至对食品安全均具有重要的研究意义。
在使用的化学农药中有40%以上含有手性结构,三唑类杀菌剂是典型代表,它们被广泛地用于农业生产、果蔬保鲜和食品工业中。烯唑醇(Diniconazole )是一种常用的三唑类农药杀菌剂[6],分子式为C15H17Cl2N3O,化学名称为(E)-(R,S)-1-(2,4-二氯苯基)-4,4-二甲基-2-(1H-1,2,4-三唑-1-基)戊-1-烯-3-醇,又名速保利,其化学结构见图1。烯唑醇是集保护、治疗、铲除作用于一体的广谱性杀菌剂,在真菌的麦角甾醇生物合成中通过抑制14α-脱甲基化而产生杀菌效果。烯唑醇的R-对映体表现出杀菌作用,而S-对映体却表现出植物调节剂的作用[7]。由于手性农药对映体功效和毒性不同,对其对映体进行拆分并测定含量很有必要。
目前,该类手性农药对映体的拆分方法研究主要集中在非手性测定,手性对映体含量的测定方法报道较少。手性拆分主要包括超临界流体色谱法[8-9]、毛细管电泳法[10-12]和高效液相色谱法[13-18]。Toribio等[8]采用衍生化直链淀粉柱,向超临界二氧化碳流体中添加不同的醇,成功地拆分了烯唑醇对映体。Ibrahim等[10]采用27 mmol/L羟丙基β-环糊精、3 mmol/L羟丙基γ-环糊精和40 mmol/L十二烷基磺酸钠(SDS)毛细管胶束电泳(MEKC)拆分了烯唑醇对映体。杨丽等[13]以衍生化β-环糊精为手性添加剂,在C8柱上拆分了己唑醇对映体,但由于流动相需消耗贵重的手性添加剂,实际应用十分有限。采用手性固定相的高效液相色谱法是拆分三唑类农药对映体更为有效的手段。Wang等[14]使用涂覆型的3,5-二甲苯氨基甲酸酯化纤维素固定相,以正己烷-异丙醇为流动相,将柱温下降至0 ℃时拆分了5种农药对映体,但溶质被洗脱的时间较长。韩小茜等[15]采用上述同类型的纤维素柱拆分了烯唑醇等对映体,考察了流动相组成和温度对分离度的影响,并计算了相关热力学参数,发现在15~25 ℃之间分离受焓控制,在25~40 ℃之间分离受熵控制。环糊精(Cyclodextrins)含有规整的手性碳和腔体,通过包结作用对许多化合物显示出手性分离能力[19]。与常用的涂覆型纤维素固定相相比,性能更稳定,自制的环糊精手性柱可节约成本,并适用于反相色谱分离模式。
本文制备了一种新型含有稳定的单脲基衍生化的β-环糊精键合的SBA-15液相色谱手性固定相,这种有序介孔固定相涡流扩散小,传质快,单脲基衍生化的β-环糊精配体对烯唑醇对映体的选择性好,用简单的甲醇-水流动相即可快速洗脱溶质,有良好的手性分离应用前景。
1实验部分
1.1仪器与试剂
Waters ZQ-2695高效液相色谱质谱联用仪,配有2996型二极管阵列检测器,色谱工作站Masslynx 4.1(美国Waters公司);5700 型傅立叶红外光谱仪( 美国Nicolet 公司) ;Vario EL Ⅲ元素分析仪(德国Elementar公司);AW-60色谱装柱机(美国Haskel公司);Milli-Q超纯水(美国Millipore公司);DS-1高速组织捣碎机(上海标本模型厂);HSC-12A氮吹仪(南京科捷分析仪器有限公司);MS-3迷你振荡器(德国IKA公司);TGL-16C高速离心机(上海安亭科学仪器厂);KQ-100E型数控超声波清洗器(昆山市超声仪器有限公司);DF-2集热式磁力搅拌器(金坛市鑫鑫实验仪器厂);ZKF-030型电热真空干燥箱(上海实验仪器总厂)。
三嵌段聚合物P123(MW~5 800)、正硅酸乙酯(TEOS)、β-环糊精(β-CD)、异氰酸丙基三乙氧基硅烷、异氰酸苯酯均购于Sigma公司;6-单乙二胺基β-环糊精(分析纯,山东滨州智源生物科技有限公司);1,3,5-三甲苯(TMB)和N,N-二甲基甲酰胺(DMF)(分析纯,阿拉丁试剂公司),DMF使用前经氢化钙除水处理;烯唑醇外消旋标准品(纯度≥99%,上海农药研究院);甲醇(MeOH,色谱纯,美国Tedia公司) ;二氯甲烷、氯化钠(国药集团化学试剂有限公司);其它试剂均为分析纯;实验用水为超纯水。Welchrom氨基柱(1 000 mg/ 6 mL,月旭材料科技上海有限公司);梨样品从市场随机购买。
1.2脲基衍生化β-环糊精键合相的制备
使用实验室前期自制的有序介孔二氧化硅SBA-15[20],以三嵌段聚合物P123作模板,正硅酸乙酯(TEOS)作硅源,1,3,5-三甲苯(TMB)为扩孔剂,在酸性条件下经水热合成法制备SBA-15。SBA-15为孔径分布约20~30 nm,粒径2.0~4.5 μm,比表面积420 m2/g的类球形颗粒。含脲基的乙二胺环糊精类键合相(UCDP)的制备路线见图2。主要合成过程如下:准确称取1.4 g干燥的6-单乙二胺基β-环糊精(A),在磁力搅拌下溶于30 mL无水DMF中,在冰浴中缓慢滴加0.3 mL异氰酸丙基三乙氧基硅烷后,室温下继续搅拌反应6 h得到产物(B)。然后,向上述溶液中加入2.5 g SBA-15,升温至110 ℃继续反应24 h,过滤后得到粗产品。将粗产品反复用DMF、甲醇、丙酮洗涤至滤液澄清,并用丙酮作为溶剂索氏提取12 h,固体于50 ℃真空干燥12 h,得到单脲基衍生化β-环糊精固定相产品(C)。
1.3色谱柱的填装
称取一定量的环糊精固定相,加入丙酮,超声使固定相均匀分散。以甲醇为顶替剂,在恒压(34.5 MPa)下,将手性固定相装填入一根不锈钢色谱柱中(4.6 mm I.D.×150 mm)。新柱用甲醇和水反复冲洗,最后用流动相平衡,直至基线稳定后进样分析。以联苯为溶质,甲醇-水(35∶65)为流动相,流速为1.0 mL/min,检测波长254 nm,测得柱效为 23 758块/m。
1.4标准溶液的配制
准确称取一定量的外消旋烯唑醇标准品,用甲醇配成1 000 μg/mL的储备溶液,即含每个对映体500 μg/mL,于4 ℃避光保存。临用前从冰箱中取出,恢复至室温,摇匀,用移液枪精密移取一定量的储备液,用甲醇稀释至刻度,得到所需浓度的烯唑醇标准溶液,超声脱气,过0.22 μm有机相滤膜后进样分析。
1.5色谱方法
衍生化β-环糊精手性色谱柱(4.6 mm I.D.×150 mm)。流动相为不同体积比的甲醇-水或乙腈,使用前经0.45 μm滤膜过滤,超声脱气10 min。流速为0.5 mL/min,检测波长为248 nm,柱温为25 ℃(298 K),进样量为10 μL。所有样品至少平行测定两次。
1.6样品前处理方法
参考相关文献进行提取、净化[21]。选取新鲜的梨试样切片,用打碎机将梨磨成果浆。分别称取梨试样10.00 g于3个50 mL离心管(1号、2号、3号)中,在1号离心管中不加外消旋烯唑醇标准样品(空白);在2号离心管中加1.0 mL 25 μg/mL的外消旋烯唑醇标准样品(样品1);在3号离心管中加1.0 mL 100 μg/mL的外消旋烯唑醇标准样品(样品2)。搅匀,作为模拟样品,并用于回收率实验。
然后分别加入20.0 mL乙腈,混匀,加入3.0 g氯化钠,振摇1 min,离心5 min,吸取乙腈液10.0 mL,在氮吹仪上吹干,加入2.0 mL甲醇-二氯甲烷(5∶95)溶解残渣。用5.0 mL甲醇-二氯甲烷(5∶95) 预淋洗氨基小柱,待液面快干时,将溶解残渣的溶液加入氨基小柱中,然后用10.0 mL甲醇-二氯甲烷(5∶95)洗脱,洗脱液经氮吹仪吹干后,用5.0 mL甲醇定容,0.22 μm有机膜过滤,待测。
2结果与讨论
2.1固定相的元素分析及红外光谱表征
对干燥后的固定相进行元素分析,结果为:C,3.24%;H,1.08%;N,0.29%。根据碳含量计算出固定相的键合量约为0.13 μmol/m2。

2.2脲基β-环糊精键合相拆分烯唑醇对映体
为提高环糊精配体的键合量和手性色谱性能,多采用全衍生化环糊精作为手性配体。但全衍生化常会导致环糊精端口部分堵塞,影响腔体的包结作用,降低拆分效果。为此,本实验制备了脲基单衍生化的β-环糊精键合固定相。脲基既是衍生化基团又是稳定的键合臂,一方面,含脲键的环糊精固定相更稳定;另一方面,脲基是典型的氢键接受体,易与烯唑醇手性碳的羟基产生氢键作用,这种端口的氢键作用有助于环糊精通过腔体包结作用识别烯唑醇的左旋体和右旋体,使得脲基衍生化环糊精固定相对烯唑醇有更好的拆分能力。采用6-乙二胺单取代β-环糊精作手性配体,通过活泼的异氰酸丙基三乙氧基硅烷偶联剂,可方便地制备稳定的脲基环糊精固定相。由于键合臂较长,环糊精配体能更有效地与烯唑醇对映体作用,采用简单、价廉的甲醇-水即可达到拆分目的。

表1 不同流动相中烯唑醇(柱温298 K)对映体的分离度
2.2.1流动相的选择烯唑醇对映体的结构很相似,拆分较困难,为了实现良好的拆分,首先需优化流动相的组成。本实验采用反相色谱,尝试用甲醇-水、乙腈-水、甲醇-醋酸三乙铵(TEAA)等作流动相,发现在脲基环糊精柱上采用常见的甲醇-水作为流动相可拆分烯唑醇。为降低测试成本,本实验选用甲醇-水为流动相,考察了不同体积比的甲醇-水流动相对分离结果的影响,结果见表1。由表1可以看出,随着甲醇比例的提高,洗脱速度加快,烯唑醇的出峰时间变短,与反相色谱分离行为相一致。与此同时,手性分离度变差,这是因为溶质的洗脱加快,溶质与固定相脲基环糊精配体之间的包结作用变差,环糊精与两对映体形成包结物的稳定性差别缩小,导致分离度下降。当甲醇-水的比例为30∶70 和25∶75时,对映体的出峰时间分别为41.82,45.23 min和75.45,80.24 min,出峰时间过长,分析速度较慢,且峰展宽,分离度未得到进一步改善。当甲醇-水的比例为40∶60时,R-和S-烯唑醇对映体的保留时间分别为14.42,15.76 min,分离度相对较大(1.56),且分析时间不超过20 min,表明流动相中含40%甲醇时可兼顾“洗脱速度较快”和“分离度较好”。因此,本实验选择甲醇-水(40∶60)作流动相,进一步优化其他色谱条件。
2.2.2流速的选择考察了流动相流速分别为0.3,0.5,0.7,1.0 mL/min时的分离效果。结果表明,较高的流速使烯唑醇对映体与色谱柱作用力不够甚至来不及作用,随着流动相而快速流出色谱柱,分离度不理想;当流速较低时,虽然有足够的时间和色谱柱作用达到理想的分离度,但大大延长了分析时间。综合考虑分离度和分析时间,选择最佳流速为0.5 mL/min。
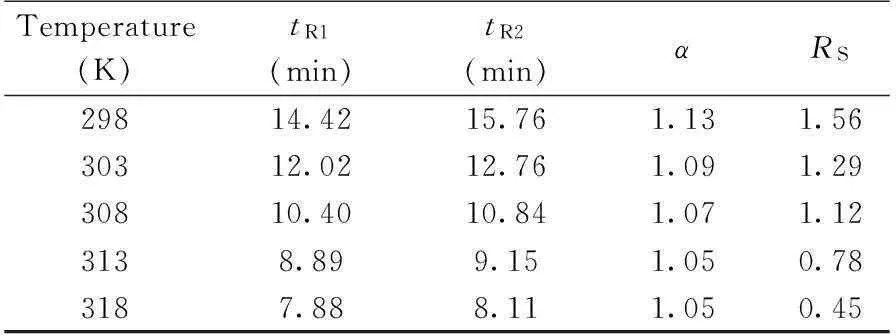
表2 不同柱温下烯唑醇对映体的分离结果
2.2.3 柱温的选择温度的变化可能引起手性固定相手性空腔中的构象发生变化,导致对映体的选择性发生变化[22]。考察了柱温分别为298,303,308,313,318 K时的分离效果,结果如表2。结果表明,在温度较低条件下,烯唑醇的两种对映体分离较好。随着温度升高,出峰时间变短,但分离度降低,虽然在298 K柱温下出峰时间较长,但时间在20 min以内,能满足高效的原则,所以选择298 K为优化的柱温。
2.2.4检测条件的选择用紫外检测器对烯唑醇标准品做多波长扫描,发现在波长200,248 nm处均有较强的紫外吸收,但由于甲醇流动相在200 nm左右也有吸收,而248 nm处则仅为烯唑醇的紫外吸收峰,因此实验设置检测波长为248 nm。
2.2.5进样量的优化分别选择进样量为1,2,3,5,10,15,20 μL以考察进样量对分离效果的影响。实验结果显示,进样量低时,流动相对烯唑醇的分离检测信号干扰较大,基线不稳定;进样量较高时,基线平稳,分离度也可满足分析要求。所以,实验选择进样量为10 μL,可减少样品对色谱柱的污染。
综合以上研究,确定优化的手性分离色谱条件为:流动相为甲醇-水(40∶60,体积比),流速0.5 mL/min,柱温298 K,检测波长248 nm,进样量10 μL。在优化的色谱条件下对烯唑醇对映体进行拆分,能达到基线平稳的较好分离(图4),同时拆分时间短,分离度为1.56,柱子选择因子为1.13。
2.3标准曲线的绘制
用甲醇适当稀释烯唑醇储备液,配制外消旋烯唑醇浓度分别为1.0,5.0,25,125,250 μg/mL标准储备液,配制每个对映体的工作浓度分别为0.5,2.5,12.5,62.5,125 μg/mL。将烯唑醇对映体标准溶液按最佳色谱分离条件进行测定,自动进样从低浓度到高浓度进行手性分离。以烯唑醇对映体的峰面积(y)为纵坐标,相应浓度(x,μg/mL)为横坐标,绘制标准曲线,并进行线性回归。结果表明,烯唑醇对映体在0.5 ~125 μg/mL范围内线性关系良好,其中R-对映体:y1=2 407.04x1+2 410.31,r1=0.999 0;S-对映体:y2=2 839.01x2+2 171.47,r2=0.999 1。以3倍信噪比计算得检出限(S/N=3)为0.02 μg/mL。
2.4回收率实验
按照“1.6”方法分别进行单个对映体浓度为1.25,5.0 μg/mL 2个加标水平的回收率实验,每个水平连续测定4次。以测得量的平均值与加入量之比计算平均回收率(表3)。结果显示,该方法有较高的回收率,梨基质中R-烯唑醇的平均回收率为92.0%~94.4%,S-烯唑醇的平均回收率为91.2%~92.8%。梨样品空白和添加烯唑醇后样品的色谱图见图5。

表3 梨样品中R- 和S- 烯唑醇的平均回收率(n=4)
2.5样品重现性实验
对添加烯唑醇的样品进行前处理后重复测定。结果显示,R- 和S- 对映体保留时间的相对标准偏差(RSD)分别为0.17%和0.44%,对映体峰面积的RSD分别为1.4%和1.7%,方法的重现性较好。
3结论
本研究以有序介孔SBA-15为基质,制备了脲基衍生化的β-环糊精手性固定相(UCDP),并将其填充于高效液相色谱柱中用以烯唑醇拆分,这种新型的方法能够较好地拆分烯唑醇对映体,分离时间约20 min,方法的重复性好,且色谱柱为自制的环糊精类色谱柱,成本较低,为手性农药的单个对映体残留量的检测提供了一种兼具快速、高效、简便、价廉等优点的新型拆分方法,为食品安全分析创造了条件。
参考文献:
[1]Pang G F.ModernAnalysisTechnologyofPesticideandVeterinaryMedicineResidues.Beijing:Science Press of China( 庞国芳.农药兽药残留现代分析技术.北京:科学出版社),2007.
[2]Tang M L,Wang D,Fu L S,Liu W P.Chin.J.Pestic.(唐梦龄,王丹,傅柳松,刘维屏.农药学学报),2011,13(4):335-340.
[3]Li Y B.StudiesontheAnalysis,EnvironmentalBehaviors,andToxicityofTypicalChiralTriazoleFungicideEnantiomers.Beijing:Chinese Academy of Agricultural Sciences(李远播.几种典型手性三唑类杀菌剂对映体的分析、环境行为及其生物毒性研究.北京:中国农业科学院),2013.
[4]Wang Q X,Qiu J,Zhou Z Q,Cao A C,Wang X Q,Zhu W T,Dang Z H.Chirality,2009,21(7):699-703.
[5]Crowell S R,Henderson W M,Kenneke J F,Fisher J W.Toxicol.Lett.,2011,205(2):154-162.
[6]Liu C,Zhu W X,Qu H Y,Li J Z,Wang H L,Guo B Y.FoodSci.(刘琛,朱文学,曲昊杨,李建中,王会利,郭宝元.食品科学),2014,35(2):154-157.
[7]Wang H L,Chen J H,Guo B Y,Li J Z.Ecotoxicol.Environ.Safety,2014,99:98-104.
[8]Toribio L,del Nozal M J,Bernal J L,Jimenez J J,Alonso C.J.Chromatogr.A,2004,1046(1/2):249-253.
[9]Zheng X Q.EnantiomerSeparationofChiralTriazoleFungicidesbySupercriticalFluidChromatography.Hangzhou:Zhejiang University of Technology(郑夏琼.手性三唑类杀菌剂在超临界流体色谱中的对映体分离研究.杭州:浙江工业大学),2013.
[10]Ibrahim W A W,Warno S A,Aboul-Enein H Y,Hermawan D,Sanagi M M.Electrophoresis,2009,30(11):1976-1982.
[11]Goetz A K,Dix D J.Toxicol.Sci.,2009,110(2):449-462.
[12]Otsuka K,Matsumura M,Kim J B,Terabe S.J.Pharm.Biomed.Anal.,2003,30(6):1861-1867.
[13]Yang L,Liao Y,Zhou Z Q,Jiang S R,Wang P.J.Instrum.Anal.(杨丽,廖勇,周志强,江树人,王鹏.分析测试学报),2004,23(5):133-135.
[14]Wang P,Liu D H,Jiang S R,Xu Y G,Zhou Z Q.J.Chromatogr.Sci.,2008,46:787-792.
[15]Han X Q,Wen X G,Guan Y H,Chen L R.Chin.J.Appl.Chem.(韩小茜,温晓光,管永红,陈立仁.应用化学),2004,21(2):140-143.
[16]Hou S C,Zhou Z Q,Qiao Z,Guo H C,Shi X Y,Wang M.Chromatographia,2003,57:177-180.
[17]Wang X S,Xu H,Wang X Y,Zhang H,Wang X Q.Agrochemicals(王雪松,徐浩,王祥云,章虎,王新全.农药),2011,50(10):711-713.
[18]Zhou Y,Li L,Lin K D,Zhu X P,Liu W P.Chirality,2009,21(4):421-427.
[19]Zhong Q Q,He L F,Beesley T E,Trahanovsky W S,Sun P,Wang C L,Armstrong D W.J.Chromatogr.A,2006,1115(1/2):19-45.
[20]Cheng B P,Li L S,Zhou R D,Li L,Zhang H F.Chem.J.Chin.Univ.(程彪平,李来生,周仁丹,李良,张宏福.高等学校化学学报),2015,36(5):872-880.
[21]Wu Y,Niu Y,Jiang R,Zhao Z D,Ma X L.J.AnhuiAgric.Sci.(吴燕,牛艳,姜瑞,赵子丹,马晓莉.安徽农业科学),2013,41(3):1089-1090.
[22]Péter A,Török G,Armstrong A W,Toth G,Tourwe D.J.Chromatogr.A,1998,828(1/2):177-190.
中美科学家合作研获单原子厚度“硼烯”
一个困扰世界凝聚态物理和材料物理界多年的难题近期被攻克。南开大学和美国阿贡国家实验室、纽约州立大学石溪分校、美国西北大学的科学家合作,首次获得了只有单原子厚度的二维硼材料——“硼烯”。该材料因其优越的电学、力学、热学属性,被科学界寄予厚望,或将成为继石墨烯之后又一种“神奇纳米材料”。相关研究发表在《科学》杂志并被重点推荐。
作为一种呈蜂巢状排列的单层碳原子结构,石墨烯是目前已知最薄、最坚硬的纳米材料,具有优良的物理化学性能,已被广泛应用于超级计算机、光子传感器等领域。继石墨烯之后,科学家希望找到更多具有优良特性的二维材料。元素硼因是碳的“近邻”而成为首要目标。然而,硼烯并非自然存在,只能人工合成。科学家对硼烯的理论结构预测已有10年之久,但从未成功合成。硼烯的制备成为国际凝聚态物理及材料物理学界公认的世界难题。
2014年,南开大学周向锋教授、王慧田教授和纽约州立大学石溪分校奥甘诺夫教授等基于进化算法结合第一性原理计算,预测了一个独特的二维硼结构。美国阿贡国家实验室、南开大学、纽约州立大学石溪分校和西北大学等研究单位共同合作,利用高真空原子溅射的方法,首次在银的表面成功“生长”出褶皱的单原子层硼烯。联合团队获得的实验结果与理论模型几乎完全符合。南开大学科研团队承担了该研究的理论计算工作。
介绍该成果的评论文章《拨开最薄硼烯的迷雾》同期刊登于《科学》杂志。《自然》杂志也刊发题为《单原子层硼烯加入二维材料俱乐部》的报道。
(信息来源:光明日报)
Separation and Determination of Diniconazole Enantiomers in Pear onβ-Cyclodextrin-based Stationary Phase by HPLCCAO Zhi-gang1,LI Lai-sheng1,2*,CHENG Biao-ping1,ZHANG Hong-fu1,ZENG Chun1,QIAO Ting1
(1.College of Chemistry,Nanchang University,Nanchang330031,China;2.Center of Analysis Testing,
Nanchang University,Nanchang330047,China)
Abstract:A new mono-ureado β-cyclodextrin-bond ordered mesoporous SBA-15 chiral stationary phase(UCDP) was prepared and packed into the column.With common methanol-water as mobile phase,the chiral pesticide diniconazole enantiomers was successfully separated by using UCDP.Effects of mobile phase composition and temperature on the chiral separation were investigated.The optimized chromatographic conditions for diniconazole enantiomers were as follows:mobile phase:methanol-water(40∶60),flow rate:0.5 mL/min,column temperature:25 ℃,detection wavelength:248 nm.Based on the above separation conditions,an HPLC method for the rapid determination of diniconazole enantiomer residues in pear was established.The pear samples were extracted with acetonitrile and purified with amino solid extraction column,and determined on UCDP in the above conditions.The good linear relationships for two enantiomers were observed in the range of 0.5-125 μg/mL.The average recoveries of the analytes in pear samples at two spiked concentration levels of 1.25 μg/mL and 5.0 μg/mL were in the ranges of 92.0%-94.4%for R-enantiomer,and 91.2%-92.8%for S-enantiomer,respectively.According to the signal-to-noise of 3(S/N=3),the detection limits of diniconazole enantiomers were 0.02 μg/mL.The UCDP preparation method is simple,and the stationary phase with urea group is stable.The developed method is rapid,accurate and reproducible,and has a good application prospect in the determination of chiral pesticide residues in fruits and vegetables.
Key words:high-performance liquid chromatography(HPLC);cyclodextrin-based chiral stationary phase;diniconazole enantiomer;chiral pesticides;food safety
中图分类号:O657.72;F767.2
文献标识码:A
文章编号:1004-4957(2016)01-0016-07
doi:10.3969/j.issn.1004-4957.2016.01.003
通讯作者:*李来生,博士,教授,研究方向:色谱分析与生化分析,Tel:0791-88304414,E-mail:lilaishengcn@163.com
基金项目:国家自然科学 (21165012);江西省自然科学 (2010GZH0089);江西省教育厅科技项目(GJJ11274)
收稿日期:2015-07-08;修回日期:2015-07-29
猜你喜欢
中国当代医药(2016年30期)2017-01-07 19:45:47
中国当代医药(2016年29期)2017-01-03 22:51:31
中国医药导报(2016年30期)2016-12-28 14:28:01
中国民族民间医药·上半月(2016年11期)2016-12-26 09:59:02
医学信息(2016年30期)2016-11-28 22:12:33
现代经济信息(2016年25期)2016-11-24 06:57:41
商场现代化(2016年26期)2016-11-21 00:20:32
中国民族民间医药·上半月(2016年10期)2016-11-19 11:36:49
新媒体研究(2016年19期)2016-11-18 19:56:49
中国科技博览(2016年18期)2016-10-19 11:03:18
