
改性细菌纤维素/羟基磷灰石复合多孔支架合成方法的研究
2016-02-16 06:19:58王巧莉奚廷斐
中国生物医学工程学报 2016年3期
南 方 王巧莉 赖 琛 奚廷斐,#*
1(温州医科大学检验医学院、生命科学学院,浙江 温州 325035)2(北京大学深圳研究院人体组织再生与修复深圳重点实验室,广东 深圳 518057)
改性细菌纤维素/羟基磷灰石复合多孔支架合成方法的研究
南 方1王巧莉2赖 琛2奚廷斐1,2#*
1(温州医科大学检验医学院、生命科学学院,浙江 温州 325035)2(北京大学深圳研究院人体组织再生与修复深圳重点实验室,广东 深圳 518057)
探讨不同合成方法对改性细菌纤维素/羟基磷灰石复合支架微观结构和性能的影响。采用原位复合法、物理混合法及生物矿化法制备改性细菌纤维素(TBC)与羟基磷灰石(HA)复合多孔支架,利用扫描电镜(SEM)、能谱分析(EDX)、X射线衍射(XRD)、傅里叶红外变换光谱(ATR-FTI)对不同方法合成的产物进行微观结构表征,同时通过力学实验确定不同支架的力学性能参数。SEM和ATR-FTI等结果表明,采用原位复合法、物理混合法及生物矿化法都可以成功地将HA复合在TBC的纳米纤维上,但是复合的机理各不相同。原位复合法中HA纳米颗粒是以螯合键的方式与TBC纳米纤维上的羧基联合,而物理混合和生物矿化法HA纳米颗粒是采用静电吸附的方式复合在TBC纤维上。XRD表明,不同方法合成的支架都出现了明显的(211)峰,但峰的形态有明显的差别。力学测试结果表明,复合后产物的力学性能也有很大的差异,采用原位复合的支架强度最低,复合后支架强度由4.67 MPa迅速减小到1.00 MPa,而用生物矿化复合的支架强度最高,复合后支架强度由4.67 MPa增加到到5.55 MPa。通过对不同方法合成的复合支架微观结构的表征和分析,为骨组织工程支架的设计提供依据。
氧化细菌纤维素;羟基磷灰石;多孔支架
引言
骨组织缺陷的修复与重造主要依赖于骨移植替代材料。胶原和羟基磷灰石是骨修复替代物中最通用的材料,因为它们的复合物能够模仿天然骨的细胞外间质。羟基磷灰石(hydroxyapatite,HA)是生物活性骨主要的无机化学成分,有良好的生物性能和骨传导性,可以促进骨细胞在材料表面生长,因此是一种常用骨修复替代材料。但是HA的力学性能较差,达不到骨组织修复材料的要求,不能单独应用于骨修复材料[1]。HA与胶原构成的复合材料也因其力学性能的原因在外压下易碎。此外胶原呈现出其他一些明显的缺陷,比如动物源性、成本及免疫原性。近年来研究发现许多材料可部分或完全的替代胶原,在骨组织修复中满足某些生理生化需求,包括明胶[2]、乳酸[3]、甲壳素[4]、壳聚糖[5]、丝素蛋白[6]及ZrO2[7]等。作为一类多糖,细菌纤维素(bacterial cellulose,BC)与胶原在纳米纤维构造方面有许多相似之处,但是BC没有免疫原性。BC是一类由细菌发酵的纤维素,包括木醋杆菌属、棘阿米巴、无色杆菌属、动胶菌和其他菌类[8]。作为一类纤维素多聚物,BC是一类由D-葡萄糖单体通过β-1,4糖苷键连接而成的线性同源多聚物[9]。与植物纤维素相比,BC纯度高,具有良好的持水性、生物相容性及力学强度和弹性模量[10]。这些特性也使BC在软骨组织工程[11]、人工小血管[12]、大鼠血管替换[13]、人造骨骼[14]、创伤敷料[15]及生物补片等方面得到广泛应用。体内外实验表明,BC作为骨再生材料可促进成骨细胞的增殖并改进骨的再生过程[16]。HA与BC构成的多孔支架可以通过多种方法获得。在许多研究里都提及的生物矿化法[17-18],用一个湿性BC模型模拟体液,包含一个与人体血浆的生理pH值和体温相同的离子浓度。物理混合法涉及到将BC的水悬浮液与制备的HA颗粒混合[19-20],它们主要通过静电吸附使HA颗粒粘附在BC表面。在原位复合法里通过在BC的水悬浮液中滴定Ca2+、PO43-和OH-形成BC与HA的复合材料[21]。然而,纤维素的主要羟基组织对磷酸钙晶体并没显示出高效的反应活性[22]。此外,羟基组织会因为超细纤维间紧密缠绕的多羟基粘合而减少。因此,HA颗粒并不会同源分布于整个BC矩阵中。为了提高HA在BC纤维上的成核作用,BC必须进行化学改性使其在纳米纤维表面上形成离子态。Okita等研究发现,BC经氧化后发生了氢键的断裂,空间结构在氧化后相对疏松[23]。还有研究表明,采用TEMPO氧化BC薄膜,可以改善材料的致密空间结构,然而仅能对其表面进行改性,对材料的整体结构改变不大[24]。我们实验室已经找到方法来改性BC来创造各类衍生物[9,25]。尽管改性反应有不少困难,主要羟基组织位点C6上仍能发生羧基化。相比于细胞羟基,BC上的羧基化活性在增强,这是因为在BC羧基化形式里羟基的均一性得以改善。
本研究中,采用原位复合、物理混合和生物矿化等3种不同的方法将HA加入氧化的细菌纤维素(TBC)中。在这些实验当中,HA的浓度相同,唯一不同的是HA纳米颗粒加入的途径。通过不同途径合成多孔支架来分析孔径大小、结构向异的差异,以探索改性BC与HA组成的复合材料能否满足竞争体制和生物先决条件,使其在骨组织工程中得到应用。
1 材料与方法
1.1 材料
(NH4)2HPO4(AR级,天津市瑞金特化学品有限公司,中国),Ca(NO3)2·4H2O(AR级,天津市大茂化学试剂厂,中国),氨水(AR级,25%,天津富宇精细化工有限公司,中国),TEMPO(AR级,Sigma公司,美国),NaClO溶液(有效氯13.4%,AR级,天津富宇精细化工有限公司,中国),NaClO2(AR级,Sigma公司,美国),细菌纤维素浆料(海南光宇生物科技有限公司,中国),磷酸盐缓冲溶液(0.05 mol/mL,pH值为6.89,上海恒远生物科技有限公司,中国)。
1.2 方法
1.2.1 细菌纤维素的氧化
TBC通过TEMPO氧化制备的[9,25]。为了在TEMPO/NaClO2/NaClO体系里获得TBC样品,首先将1 g BC加入300 mL磷酸盐缓冲液中。然后加入0.1 mmol TEMPO和17 mmol NaClO2,将2 mL NaClO加入100 mL磷酸缓冲液中混合均匀,然后将混合液加入BC的悬浮液中。最后将混合溶液快速密封并放在磁力搅拌器上65℃恒温搅拌30 h。关掉磁力搅拌器,加入10 mL乙醇终止反应。将反应产物放入高速离心机中离心,弃掉上清冲洗壁上的沉淀物再离心,反复3次后放入低温冰箱,冷冻干燥48 h后得到干燥的氧化纤维素样品。
1.2.2 细菌纤维素氧化后羧基含量的测定
TBC中羧基含量的测定可以采用电导率滴定法[26]。称取0.2 g干燥后的TBC样品,放入盛有70 mL水的烧杯中,加入3 mL 0.01 mol/L的Nacl溶液,在高速搅拌机下分散成匀浆。再向上述体系加入0.1 mol/L的HCl溶液,调节溶液的pH值到2.5~3.0之间。然后以0.04 mol/L的NaOH溶液作为标准的滴定液,用电导率滴定仪对上述体系进行滴定测试。滴定中可能会有少部分的NaOH参与到副反应中,但大部分的NaOH是参与到了与羧基的中和反应中。因此,可根据NaOH的消耗量来计算羧基含量[27]。羧基含量的计算公式为
(1)
式中:V2是右边等当点NaOH溶液的体积,mL;V1是左边等当点NaOH溶液的体积,mL;C是NaOH溶液的浓度;W是干燥的TBC的质量。
1.2.3 氧化细菌纤维素和羟基磷灰石多孔支架的制备
对于原位复合法,分别制备0.5 mol/L的(NH4)2HPO4和1 mol/L的Ca(NO3)2溶液,放入滴定管中。取1 g干燥的TBC样品在室温下放入去离子水中并在25 000 r/min搅拌器下搅拌,形成悬浮的浆料。(NH4)2HPO4和Ca(NO3)2溶液在室温下通过滴定管同时缓慢的加入到搅拌速率为5 000 r/min的TBC浆料中,滴定管的速率保持在4 mL/min,利用NaOH(0.5 M)保持溶液的pH值在11以上。反应结束后将混合体系放入真空干燥器中1 h除去气饱。然后转入聚四氟乙烯模具(12.5 cm×8.5 cm×2.0 cm,64孔)中,放入-20℃低温冰箱中冷冻24 h。再将冷冻的样品放入冻干机中,冷冻干燥48 h,获得多孔支架。
在物理混合法中,0.5 mol/L(NH4)2HPO4和1 mol/LCa(NO3)2溶液共同倒入密封反应釜中,在120℃下反应24 h,自然冷却,过滤沉淀,用去离子水清洗3遍,75℃的烘箱中干燥24 h,获得平均长度500 nm的针状HA颗粒。并如上述方法制备TBC悬浮液。在搅拌速率为1 000 r/min的TBC悬浮液中加入HA颗粒,8 h后,停止反应,如上述方法,制备TBC/HA支架。
生物矿化法制备多孔支架,分别配制1 mol/L CaCl2和0.5 mol/LNaH2PO4的矿化液。将柱形TBC(高10 mm,直径5 mm)的支架分别依次浸入CaCl2和NaH2PO4溶液中,在37℃、转速50 r/min条件下震荡30 min,在每次支架从矿化液中取出时,用去离子水冲洗,用滤纸吸干多余的液体,再放入另一种矿化液中,进行3~8次循环。最终样品在37℃烘箱中干燥3 d。
1.2.4 多孔支架场发射扫描电镜(SEM)检测
采用扫描电镜(MIRA3型,TESCAN, 捷克)观察样品的表面形态,在观察前先对样品进行喷金处理,支架样品一般会进行2次喷金处理。比较3种不同的方法制成的多孔支架的表面形态。观察过程中对生物矿化制作的支架样品选择晶体相对集中的位置,并对此区域的物质用能谱分析仪(Genesis Apollo X/XL型,EDAX,美国)进行能谱分析。
1.2.5 多孔支架的X射线衍射(XRD)检测
使用X-衍射仪(D/max-2500/PC型,Rigaku,日本),对TBC、HA、不同方法制备的TBC/HA样品进行测试。测试的条件:CuKa射线、Ni滤波、40 mA电流,扫描的范围2θ=10°~40°。
1.2.6 多孔支架傅里叶红外变换光谱(FTIR)测定
采用光谱仪(Nicolet 6700型,Thermo, 美国)来测量冻干的复合支架样品的ATR-FTIR光谱,采样器进行32次扫描,分辨率为4 cm-1,扫描范围为1 100~1 700 cm-1。
1.2.7 多孔支架力学强度的测试及相对密度
利用万能力学试验机(Z005型,Zwick,德国)测量支架材料的力学性能。以压缩速度5 mm/min,压缩比100%为控制条件。比较3种不同的方法制得的纳米复合材料的压缩杨氏模量。
相对密度和杨氏模量关系密切,它对支架的硬度和强度有明显的影响,计算方式为
(2)
式中,E*和Es分别表示多孔支架和疏松材料的杨氏模量,ρ*和ρs分别表示多孔固体和疏松材料的密度;C是一个常数,在Oscar′s的实验里表明它与孔的规律性成反比[28]。C值的增加表示支架中孔的分布更加随意及结构向异性变大,而不同的多孔体系指数不同。
2 结果
2.1 SEM分析
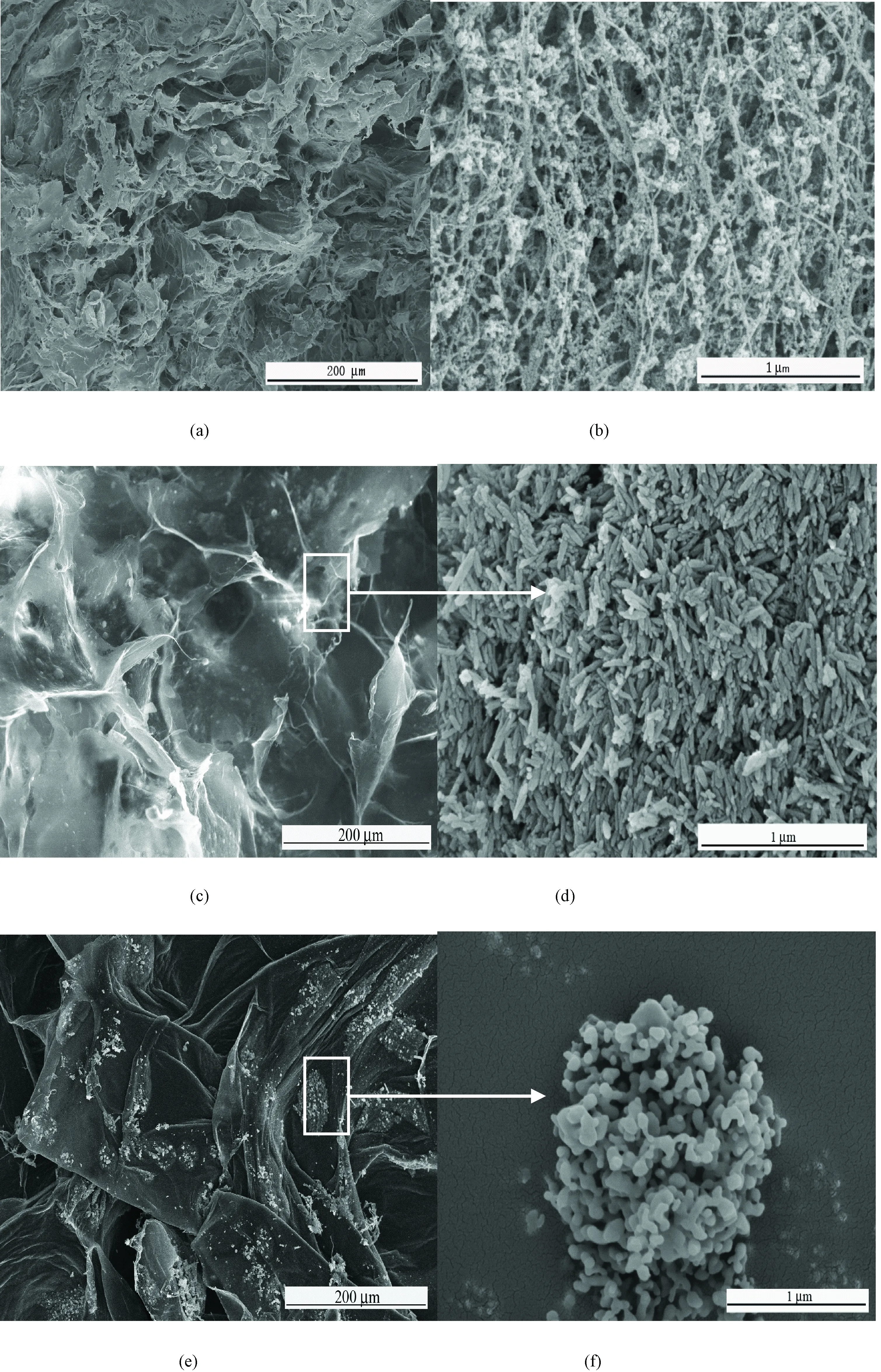
图1 不同方法制备的多孔支架的电镜交联区域图片。(a)和(b) 原位复合法按照TBC∶HA=1∶1.5制成多孔支架及其局部放大;(c)和(d)物理混合法的质量比是TBC∶HA=1∶1.5及其局部放大;(e)和(f)生物矿化法中TBC多孔支架6次循环浸入CaCl2和NaH2PO4溶液及其局部放大Fig.1 Cross-sectional SEM images of sponges prepared under different methods.(a) and (b) Sponges prepared by in situ formation with a weight ratio of TBC:HA=1∶1.5 and local enlarged image; (c)and(d) Sponges prepared by physical mixing with a weight ratio of TBC:HA=1∶1.5 and local enlarged image; (e) and (f) Biomineralized TBC sponges after 6 cycles of alternate soaking in calcium and phosphate solutions and local enlarged image
不同方法制备支架的扫描电镜如图1所示。在不同的TBC多孔支架上HA纳米颗粒的尺寸和形态不同。从支架的截面图可以看到,采用原位复合法制备的多孔支架(见图1(a)),孔径较小,孔道较深,孔隙率也相对较大。从局部放大图(见图1(b))可以清楚地看到,TBC的纳米纤维较为疏松分散,形成疏松的网络结构,HA的纳米颗粒呈球形,附着在单根纳米线或者纳米束上。采用物理混合法制备的多孔支架(见图1(c))空隙率明显降低,孔道不贯通,有明显的白色点状沉淀附着在支架中,局部放大图(见图1(d))显示点状沉淀是针状HA纳米颗粒的聚集体。针状是水热法合成HA的典型形态。矿化法制备的支架(见图1(e))由于反复浸渍循环,孔道出现坍塌,有较多的白色固体沉淀在支架中,局部放大图(见图1(f))显示HA颗粒呈珊瑚状堆积。
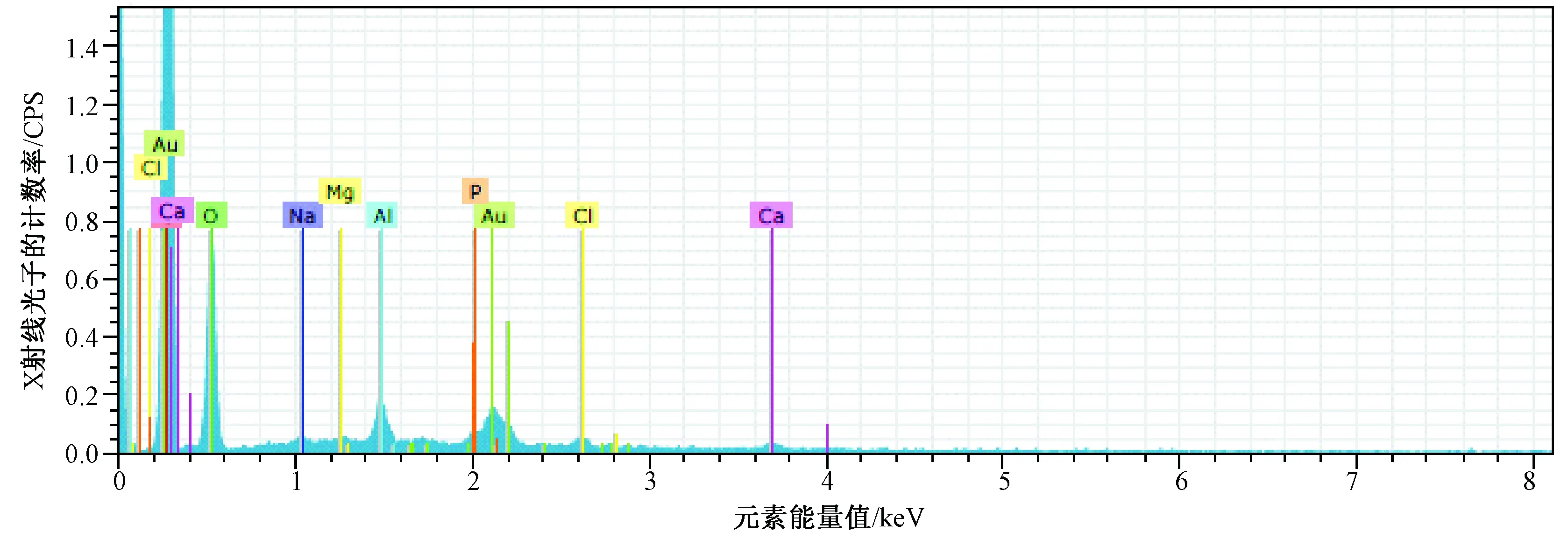
图2 TBC/HA复合材料矿化7 d的EDX图谱Fig.2 Spectra of TBC/HA composite mineralization for 7 days
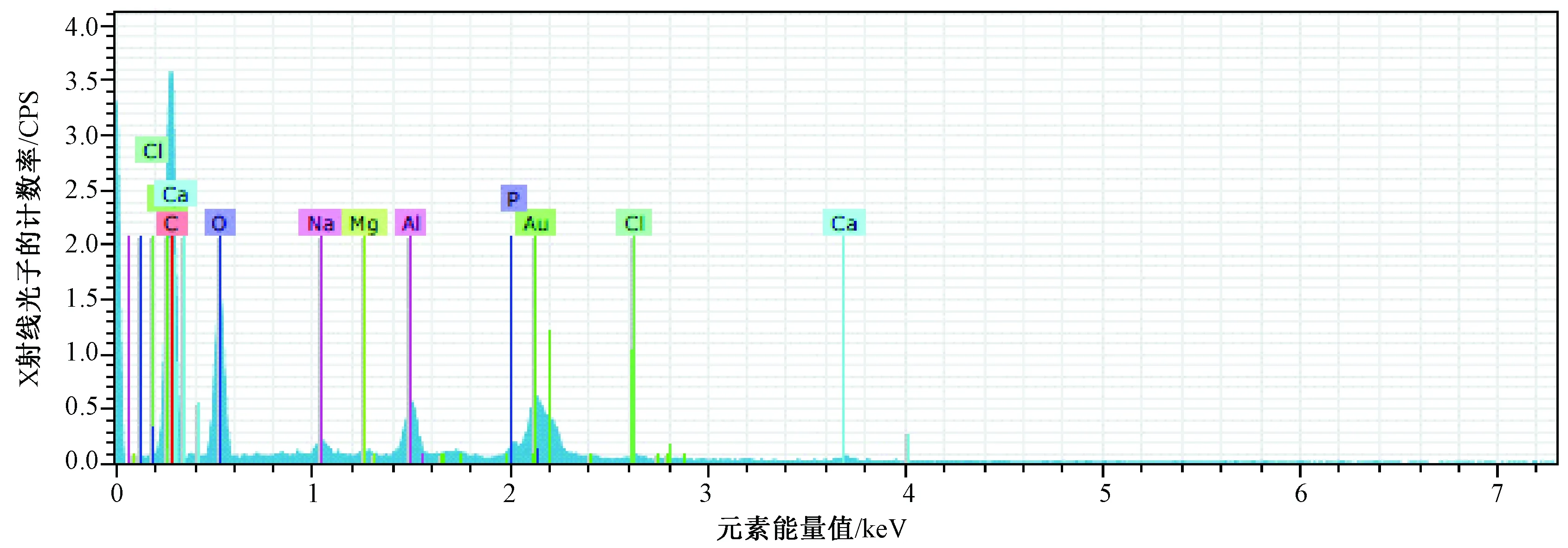
图3 TBC/HA复合材料矿化21 d的EDX图谱Fig.3 EDX spectra of TBC/HA composite mineralization for 21 days
2.2 EDX分析
在电镜观察矿化法制备的TBC/HA复合支架时,寻找晶体相对聚集的地方做能谱分析。EDX图谱中各原子比例见图2、3,本实验选择的是矿化7和21 d的两组支架所做的能谱分析。表1和表2显示了支架中含有碳、氧、铝、钙、磷等元素。其中,矿化7d的钙磷比例为1.28,矿化21 d的钙磷比例为1.55。羟基磷灰石中钙磷的比例为1.67,生物矿化法中的钙磷比相比羟基磷灰石中的钙磷比略低些。

表1 TBC/HA复合材料的原子比列Tab.1 The Element ratio of TBC/HA composite
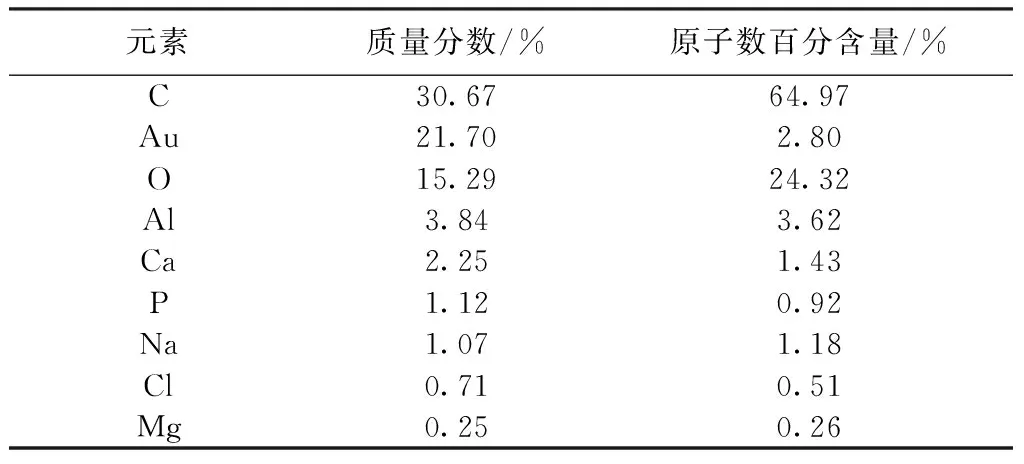
表2 TBC/HA复合材料的原子比列Tab.2 The Element ratio of TBC/HA composite
2.3 XRD分析
各种支架样品的XRD图谱如下所示。图4中曲线A是纯HA的衍射图谱,尖锐的峰型表明了水热法合成的HA具有较高的结晶度,31.5、32.9、34.0度分别对应了(211),(112),(202)面。3种复合方法制备的复合材料具有类似的XRD衍射图形,同时出现了(211)面的衍射峰。原位复合法制备的材料(见图4中曲线B),(211)峰是非常明显的无定形态宽峰,说明了在此体系中出现的HA颗粒基本属于无定形态。但非常明显的区别是,矿化体系中获得的复合材料(见图4中曲线C),(211)峰相对尖锐,表明体系中存在的HA的结晶程度比原位法要强。物理混合法制备的TBC/HA复合材料出现了比较尖锐的(211)峰(见图4中曲线D),表明了体系中存在着晶型较好的HA。TBC的XRD图谱(见图4中曲线E)展示了典型的纤维素晶体结构的图形,在14.4和22.5处出现明显的峰值,分别对应于(112)和(002)面。这与纯BC的图谱[9]基本上没有差异。

图4 TBC/HA复合材料X射线衍射图(A-纯HA;B-TBC∶HA=1∶1.5(原位复合法);C - TBC:HA6次循环(生物矿化法);D-TBC:HA=1∶1.5(物理混合法);E-TBC)Fig.4 X-ray diffraction patterns of TBC-HA composites(A-Pure HA;B-TBC∶HA=1∶1.5(in situ formation);C-TBC:HA 6 cycles(biomineralization);D-TBC∶HA=1∶1.5(physical mixing);E-TBC)
2.4 FTIR分析
各种样品的红外反射图如图5所示。因为主要是研究COO-基团与Ca2+之间相互作用的形态,所以本研究集中于从1 700~1 100 cm-1中外区域部分的峰值变化。对于生物矿化法的样品(见图5中曲线A),COO-很难识别并分裂成许多小峰,说明在生物矿化的过程中,COO-与Ca2+发生的化学反应比较复杂。物理混合法的图谱中COO-振动峰消失(见图5中曲线B),但是在1 481~1 431 cm-1范围内出现较宽的峰,证明有较弱的新键产生。在1 481和1 431 cm-1处新出现的峰,表明了采用原位法制备的复合物中形成了新的COO-Ca螯合键(见图5中曲线C)。在1 552和1 417 cm-1处的峰是TBC中COO-的振动峰(见图5中曲线D)。

图5 各种样品的ATR-FTIR图谱(A-生物矿化法制备的TBC/HA(6次循环);B-物理混合法制备的TBC/HA(TBC∶HA=1∶1.5);C-原位复合法制备的TBC/HA(TBC∶HA=1∶1.5);D-TBC)Fig.5 ATR-FTIR spectra of various samples (A-TBC/HA prepared by biomineralization(6 cycles);B-TBC/HA prepared by physical mixing (TBC∶HA=1∶1.5); C-TBC/HA prepared by in situ formation(TBC∶HA=1∶1.5); D-TBC)
2.5 力学性能分析

图6 不同方法制备样品的杨氏模量。(a)原位复合法;(b)物理混合法;(c)生物矿化法Fig.6 Young′s modulus of samples prepared by different methods. (a) In situ formation; (c) Physical mixing; (d) Biomineralization
各种样品的力学性能如图6所示。不同的复合方法,复合材料的力学性能有很大不同,可见HA的形态对材料的力学性能有很大的影响。原位复合法制备的多孔支架,HA的出现会引起杨氏模量显著减小(见图6(a)),这表明在TBC的网络结构中HA的出现会削弱力学特性。在物理混合法当中,HA颗粒的加入在低水平区域内(质量比TBC∶HA=1∶0.75,图6(b))使得杨氏模量轻微的增加。当质量比增加到1∶1.5时,杨氏模量明显减小。在图6(c)中,能看到生物矿化法合成支架的杨氏模量随着HA比例的增高有轻微增加的趋势。
相对密度是指多孔固体与疏松材料的密度比值(ρ*/ρs)[28],对于多孔材料来说它是一个重要的参数,与力学强度也有较大的关系。
图7显示了不同样品的相对密度。原位复合法和物理混合法做了4个浓度梯度的样品,矿化法对应于3次循环、5次循环、6次循环和8次循环。相对密度的变化趋势与弹性模量比较相似。纯BC材料因为微观结构致密因而有高的相对密度。原位复合法制备的多孔支架结构疏松,孔隙率高,因此密度低。物理合成法中HA含量增加,密度降低,呈现一个最小值后然后开始增加。采用生物矿化法制备的支架强度明显比另两种方法要强很多,甚至比纯BC支架都要强。从以上测试结果可以得出结论,HA与TBC的复合方式以及HA的微观形态对复合材料的微观结构和宏观性能都有较大的影响。

图7 不同方法制备的各种样品的相对密度Fig.7 The relative density various samples prepared by different methods.
2.6 羧基含量的测定
BC经过氧化后,C6上的羟基最终被氧化成了羧基,通过电导率滴定法根据消耗的NaOH的量来计算氧化后细菌纤维素中羧基的含量。表3所示是选取的3组数据计算的羧基的含量。可以看出,每组测量因为氢氧化钠消耗量的差异,羧基含量稍微有些差别。0.2 g的TBC样品中,羧基含量平均值为0.67 nmol/g。

表3 羧基的含量Tab.3 The content of carboxy
3 讨论
BC经过TEMPO/NaClO2/NaClO氧化体系进行改性后,C6上的-OH先被选择性的氧化成-CHO,在进一步的氧化成-COOH基团(见图8(a)、(b))。氧化后的TBC与BC在结构上比较而言,亲电基团增多,氢键作用减弱,结构由致密变得相对疏松。

图8 不同方法制备的多孔支架的内部反应原理。(a)纯BC;(b)TBC;(c)生物矿化;(d)物理混合;(e)原位复合。Fig.8 The internal reaction mechanism of different methods for preparation of porous scaffolds.(a) Pure BC;(b)TBC; (c) Biomineralization; (d) Physical mixing; (e) In situ formation.

XRD图显示了各种复合方法如何影响材料的晶体结构。对于TBC而言典型的I型纤维的衍射峰是在2θ图里14.4和22.5处出现的峰。纯的HA纳米颗粒在32.9处也出现典型的晶型峰(211),同时COO-分裂形成了其他3个小的新型峰。原位复合法里,晶型图的线条比较粗糙,(211)处的峰呈无定形态的宽峰。这说明原位法合成的多孔支架的HA晶体结构差。物理混合法构成的复合材料的晶型图与TBC的相似,说明此法构成的支架结晶度和密度高。由于HA颗粒在TBC中不均匀的分布,在该图谱中没有相应的HA峰出现。生物矿化法也没有相应的HA峰的产生,但是在32处出现了一个宽的峰,说明TBC和HA可以共存的。
通过观察FT-IR图可知,TBC在1552/1417两处出现了明显的羧基峰。对比3种不同复合方法的红外图谱可知,在原位复合当中,由于TBC上的COOH与HA中的Ca2+形成了COOCa,所以在1431/1481处产生新的峰。物理混合法里,与TBC图谱比较而言,图谱变化不大说明TBC与HA反应微弱。在矿化反应中出现了许多的小峰,而且羧基峰很难识别,这是由于TBC的保护使其活性减弱。3种方法构成的复合支架的红外图都表明,HA晶体被成功地结合到TBC空间结构中。
3种方法构成的多孔支架的杨氏模量图变化很明显,说明HA的形态对多孔支架力学性能有显著的影响。在原位法里,杨氏模量变化很明显,有近75%的减小,说明HA颗粒会削弱纤维素之间的力学强度。物理法中,只有当TBC与HA比例从1∶1.5到1∶3时,杨氏模量迅速减少。这说明,小比例的HA的加入对纤维的力学性能影响不大,但超过这个比例,力学性能会被严重削弱。矿化法里,随着HA的不断加入,力学性能基本不变。这些结果表明,HA颗粒的晶型结构对力学性能有显著影响。
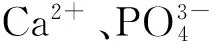
4 结论
对改性的细菌纤维素与羟基磷灰石纳米颗粒,采用原位复合法、物理混合法及生物矿化法等3种不同方法构成的多孔支架,进行SEM、EDX、XRD、FTIR、力学性能及相对密度进行测试分析。3种方法都可以使HA颗粒成功的结合在TBC上。对比3种方法形成的支架对微观结构的影响,原位复合法通过螯合作用和共价键使支架的孔径变大、结构疏松分布不规律、支架的力学强度最小。生物矿化法COOH活性被保护,反应复杂,有珊瑚状致密结构产生、支架的力学强度最高。物理混合法通过静电作用使支架孔径逐渐被堵、孔的横截面变大、微观结构整齐、支架的力学强度介于两者之间。比较而言,螯合作用和静电作用在TBC与HA反应中占主导地位。所构成的多孔支架基本上满足了竞争体制要求,可用于骨组织修复材料。不过本实验以细菌纤维素作为基础材料,通过氧化改性再与羟基磷灰石进行复合,制备骨修复材料的研究只限于微观结构的表征分析,如在临床上得以应用,还需进一步的动物实验。
[1] White AA, Best SM. Hydroxyapatitie-Carbon nanotube composites for biomedical application: A review [J]. Int J Appl Ceram Technol, 2007,4(1):1-13.
[2] Chang MC, Ko CC, Dounglas WH. Preparation of hydroxyapatite-gelatin nanocomposite[J]. Biomaterials, 2003,24(17):2853-2862.
[3] Ignjatovic N, Uskokovic D. Synthesis and application of hydroxyapatite/polylactide composite biomaterial [J]. Appl Surf Sci, 2004, 238(1-4):314-319.
[4] Ge ZG, Baguenard S, Lim LY, et al. Hydroxyapatitechitin materials as potential tissue engineered bone substitutes l [J]. Biomaterials, 2004, 25(6):1049-1058.
[5] Zhang L, Li YB, Yang AP, et al. Preparation and in vitroinvestigation of chitosan/nano-hydroxyapatite compositeused as bone substitute materials[J]. J Mater Sci Mater Med, 2005,16 (3):213-219.
[6] Cai Zengxiao, Mo Xiumei, Zhang Kuihua, et al. Fabrication of chitosan/silk fibroin composite nanofibers for wound-dressing applications [J]. International Journal of Molecular Sciences, 2010, 11(9):3529-3539.
[7] Rapacz-Kmita A, Slosarczyk A, Paszkiewicz Z. Mechanical properties of HAp-ZrO2 composites [J]. J Eur Ceram Soc,2006,26(8):1481-1488.
[8] 黄建文, 徐月敏. 细菌纤维素在组织工程中的应用[J]. 中国组织工程研究,2014,18(3):420-425.
[9] Chen Lai, Shujiang Zhang, Xuanchen Chen,et al. Nanocomposite films based on TEMPO-mediated oxidized bacterial cellulose and chitosan [J].Cellulose,2014, 21:2757-2772.
[10] Retegi A, Algar I, Martin L, et al. Sustainable optically transparent composites based on epoxidized soy-bean oil (ESO) matrix and high contents of bacterial cellulose (BC) [J]. Cellulose, 2012, 19(3):103-109.
[11] Svensson A,Nicklasson E,Harrah T,et al.Cellulose as a potential scaffold for tissue engineering of cartilage [J]. Biomaterials,2005,26: 419 -431.
[12] Carl JM, Bo R, Aase B, et al. Small calitre biosynthetic cellulose blood vessels: 13-months patercy in a sheep model [J]. Scandonavian Cardiovascular Journal, 2012, 46(1):57-62.
[13] Klemm D, Schumann D, Udhardt U, et al.Bacterial synthesized cellulose-aritificial blood vessels for microsurgery. [J]. Prog Polym Sci, 2001;26:1561-1603.
[14] Hutchens SA, Benson RS, Evans BR, et al. Biomimetic synthesis of calcium-deficient hydroxyapatite in a natural hydrogel [J]. Biomaterials,2006,27(26): 4 661-4670.
[15] Wiegand C, Elsner P, Hiple UC. Protease and ROS activities influenced by a composite of bacterial cellulose and collagen type I in vitro [J]. Cellulose, 2006, 13 (6):689-696.
[16] Nurlidar F, Budianto E, Darwis D, et al. Hydroxyapatite deposition on modified bacterial [J]. Cellulose Matrix Macromol Symp. 2015, 353, 128-132.
[17] Zimmermann KA, LeBlanc JM, Sheets KT, et al. Biomimetic design of a bacterial cellulose/hydroxyapatite nanocomposite for bone healing applications[J].Materials Science and Engineering C, 2011,31:43-49.
[18] Wan YZ, Hong L, Jia SR, et al. Synthesis and characterization of hydroxyapatite-bacterial cellulose nanocomposites [J].Composites Science and Technology, 2006, 66:1825-1832.
[19] Fan Xiaoxia, Zhang Tingting, Zhao Zhitong,et al.Preparation and characterization of bacterial cellulose microfiber/goat bone apatite composites for bone repair[J]. Apllied Polymer Science,2013,129(2):595-603.
[20] Guerra GD, Cristallini C, Urciuoli P, et al. Hydroxyapatite/gelatin/gellan sponges as nanocomposite scaffolds for bone reconstruction[J]. Materials in Medicine,2012,23(1): 51-61.
[21] Luo Honglin, Xiong Guangyao, Zhang Chen, et al. Surface controlled calcium phosphate formation on three-dimensional bacterial cellulose-based nanofibers[J].Materials Science and Engineering C, 2015, 49:526-533.
[22] Gonzalez M, Hemandez E, Ascencio JA, et al.Hydroxyapatite crystals grown on a cellulose matrix using titanium alkoxide as a coupling agent[J]. J Mater Chem,2003,13: 2948-2951.
[23] Okita Y, Saito T, Isogai A. Entire surface oxidation of various cellulose microfibrils by TEMPO-mediated oxidation [J].Biomacromolecules, 2010,11(6):1696-1700.
[24] Lu Chuan, Chen Shiyan, Zheng Yi, et al. TEMPO-mediated oxidation of bacterial cellulose in buffer solution [C]//Han Yafang. Materials Science Forum. Swizerland: Trans Tech Publications Inc,2014,789:90-94.
[25] Lai Chen, Sheng Liyuan, Liao Shibo,et al. Surface characterization of TEMPO-oxidizedbacterial cellulose[J]. Surf Interface Anal,2013, 45: 1673-1679.
[26] Toei K, Kohara T. A conductometric method for colloid titrations [J]. Anal Chimica Acta,1967, 83: 59-65.
[27] Sun Bin, Gu Chunju, Ma Jinhong, et al. Kietic study on TEMPO-mediated selective oxidation of regenerated cellulose [J]. Cellulose, 2005, 12(1): 59-66.
[28] Sotomayor OE, Tippur HV. Role of cell regularity and relative density on elastoplastic compression response of 3D open-cell foam core sandwich structure generated using Voronoi diagrams [J]. Acta Materialia, 2014,78: 301-313.
Research on the Synthesis of Modified Bacterial Cellulose/Hydroxyapatite Sponges
Nan Fang1Wang Qiaoli2Lai Chen2Xi Tingfei1,2*
1(SchoolofLaboratoryMedicineandLifeScience,WenzhouMedicalUniversity,Wenzhou325035,Zhejiang,China)2(ShenzhenkeyLaboratoryofHumanTissueRegenerationandRepair,ShenzhenInstituteofPekingUniversity,Shenzhen518057,Guangdong,China)
The purpose of this study is to investigate three kinds of preparation methods for bacterial cellulose and hydroxyapatite sponges and compare the microstructure and characteristics.The modified bacterial cellulose (TBC) and hydroxyapatite (HA) sponges were fabricated byinsituformation, physical mixing orbiomineralization. Samples prepared by the different methods were characterized using scanning electron microscopy (SEM), energy spectrum analysis(EDX),X-ray diffraction (XRD), Fourier IR transform spectroscopy (ATR-FTI). Furthermore mechanical performance of the sponges prepared with different parameters were tested as well.Experimental results showed that by theinsituformation, physical mixing orbiomineralization HA was successfully deposited on TBC nanofibers, but mechanisms were different.By theinsituformation method, HA nanoparticles took the form of chelate keys associated with TBC nano fibers on the carboxyl. In the physical mixing orbiomineralization methods, HA nanoparticles became electrostatically adsorbed on TBC nanofibers.XRD results showed that there were obvious (211) peaks in the scaffolds synthesized by different methods, but there were significant differences in the morphology of the peaks.Mechanical results showed that microstructure and mechanical properties of sponge also had very big difference with different composite methods. By theinsituformation method, the scaffold minimum intensity was reduced from 4.67MPa to 1.00MPa,while by the biomineralization, scaffold maximum intensitywas increased from 4.67MPa to 5.55MPa. Through the analysis of the microstructure and characterization of the scaffolds,our study could provide the basis for their application to bone tissue engineering.
oxidation of bacterial cellulose; hydroxyapatite; sponge
10.3969/j.issn.0258-8021. 2016. 03.011
2015-09-25, 录用日期:2016-01-22
深圳市基础研究项目(JCYJ20140419114548513)
R318
A
0258-8021(2016) 03-0330-010
# 中国生物医学工程学会会员(Member, Chinese Society of Biomedical Engineering)
*通信作者(Corresponding author), E-mail: xingtingfei@pku.edu.cn
猜你喜欢
幼儿100(2024年19期)2024-05-29 07:43:34
建材发展导向(2022年24期)2022-12-22 07:44:36
环境卫生工程(2021年4期)2021-10-13 06:52:26
天然产物研究与开发(2018年5期)2018-06-13 03:23:54
应用化工(2014年10期)2014-08-16 13:11:29
应用化工(2014年7期)2014-08-09 09:20:23
丝绸(2014年5期)2014-02-28 14:55:12
丝绸(2014年3期)2014-02-28 14:54:52
河南科技(2014年12期)2014-02-27 14:10:29
华东理工大学学报(自然科学版)(2014年6期)2014-02-27 13:49:40
