
三江平原沼泽湿地垦殖及自然恢复对土壤细菌群落多样性的影响
2016-02-07 00:53蔡体久琚存勇唐庆明
生态学报 2016年22期
徐 飞, 蔡体久, 杨 雪, 琚存勇, 唐庆明
1 东北林业大学林学院, 哈尔滨 150040 2 黑龙江省农垦科学院, 哈尔滨 150038 3 呼伦贝尔市林业局, 呼伦贝尔 021008
三江平原沼泽湿地垦殖及自然恢复对土壤细菌群落多样性的影响
徐 飞1, 2, 蔡体久1,*, 杨 雪1, 2, 琚存勇1, 唐庆明3
1 东北林业大学林学院, 哈尔滨 150040 2 黑龙江省农垦科学院, 哈尔滨 150038 3 呼伦贝尔市林业局, 呼伦贝尔 021008
为量化垦殖对我国东北沼泽湿地的影响程度,于2015年6月基于Illumina Miseq PE300第二代高通量测序平台,对黑龙江省洪河国家级自然保护区的原始沼泽、耕地、退耕湿地的土壤细菌16S rRNA基因高变区域进行测序,并分析沼泽湿地土壤细菌群落多样性和结构组成与垦殖的关系。结果表明:共获得358737条修剪序列且被划分为36个已知的菌门,在11个主要的土壤细菌门类中 (相对丰度>1%),芽单胞菌门 (P<0.01)、拟杆菌门 (P<0.01)、厚壁菌门 (P<0.01) 和绿菌门 (P<0.01) 在不同生境类型样地差异显著,土壤细菌群落alpha多样性由高到低排序依次是:耕地、退耕湿地、原始沼泽。结合相关性分析和冗余分析可以证明,长期的耕作,特别是旱田耕作对沼泽湿地土壤细菌群落组成产生显著的影响,土壤pH、含水量、全碳、有机碳、可溶有机碳、碱解氮、微生物量碳、微生物量氮对土壤细菌的多样性和群落组成产生影响。总之,研究结果发现自然恢复可以显著促进土壤细菌群落多样性的恢复,但是沼泽湿地土壤细菌群落结构一旦被垦殖干扰改变将很难恢复到原始状态,强调了有效地利用土壤细菌群落对维护土壤生态系统平衡具有重要的意义。
沼泽;垦殖;土壤细菌;α多样性;群落结构;高通量测序
中国东北部是沼泽湿地 (Marsh) 的主要分布区之一,也是黑土的原生地。由于特殊的气候条件和地理位置,使土壤具有腐殖质含量高、微生物数量和种类较少的特点。因此,其生态环境虽然最适合农业开发,但具有脆弱易破坏性[1- 2]。在全球气候变暖和人类大面积垦湿造田的综合影响下,中国东北沼泽湿地出现了不同程度的退化。沼泽湿地生态系统正常的水循环、有机物和无机物的循环过程受到严重影响,土壤变薄、变贫瘠,地下水位降低,沼泽湿地的土壤微生物的群落结构和组成随之发生改变[3- 5]。
近年来,这种现象逐渐引起中国政府及科研人员的高度关注。有关湿地垦殖的影响[6]、湿地污染的处理[7]、湿地微生物参与的碳氮循环等方面成为了研究热点[8- 9]。人类逐渐认识到土壤微生物是土壤生态环境中最敏感的生物,并且不断探索通过控制土壤微生物促进土壤环境修复的方法[10]。而细菌在土壤微生物中数量最多、分布最广,是参与土壤有机质分解和矿化过程的主要门类,在化学物质循环、降解污染和修复生态环境方面起着重要作用[11]。其群落结构与组成能够反映出土壤养分变化乃至环境变化带来的影响,并能直接影响土壤功能的发挥[12]。因此,研究人员对土壤细菌的种类和功能进行更详细的划分,对多种土壤细菌分解有机质、固氮、控制植物生长和降解有机农药的调节机制展开了深入的研究[13- 15]。
然而,传统的观察和分离方法由于仪器的分辨率和大量的微生物不能够培养带来的局限性等因素,很难准确的揭示细菌的群落结构、功能及多样性。随着科技的进步,分子生物技术成为科学家们探索微生物个体、群体间作用及其与环境关系的重要手段[16- 17]。不可培养的土壤细菌通过基因组信息能够被辨别、丰度可以被比较、计算,因此全面了解土壤细菌群落状态成为了现实[18]。本文以垦殖及自然恢复下的沼泽湿地为研究对象,基于新一代高通量基因测序技术,通过分析土壤细菌的种类以及它们之间的相对丰度和进化关系,探讨土壤细菌多样性与土壤环境因子之间的内在联系,揭示垦殖对中国东北沼泽湿地土壤环境带来的影响,对于研究环境治理和微生物资源利用有着重要的现实意义,为中国东北沼泽湿地的保护和利用提供科学依据。
1 实验方法
1.1 研究地区概况
试验地位于三江平原腹地黑龙江省洪河国家级自然保护区(47°42′38″—47°52′00″N, 133°34′38″—133°46′29″ E),总面积为21836 hm2,属于湿地生态系统类型的保护区。海拔一般在58—61 m之间,温带湿润气候,1月份平均温度为-23.4℃左右,7月份平均温度在22.4℃左右,年平均气温约为1.9℃。多年平均降雨量为585 mm,多年平均蒸发量为1166 mm,多年平均无霜期131 d左右,冻融期从10月中旬至次年的5月中旬,沼泽植被冻层深80—160 mm。保护区内的绝大部分植被仍保持原始状态,以草本沼泽植被和水生植被为主,间有岛状林分布。主要植被有毛果苔草 (Carexlasiocapa)、漂筏苔草 (Carexpseudocuraica)、乌拉苔草 (Carexmeyeriana)及狭叶甜茅 (Glyceriaspiculosa)、小叶章 (Deyeuxiaangustifolia)等[19]。
1.2 样地的设计与土壤样品的采集
2014年9月,根据洪河国家级自然保护区内沼泽湿地和实验田的分布情况,共设置了6块实验样地。其中,在保护区缓冲区设置2块原始沼泽湿地类型样地 (PM),分别命名为小叶章沼泽 (Site 1, S1)、乌拉苔草沼泽 (Site 2, S2);在保护区实验区设置2块耕地类型样地 (Cultivated land, CL),分别命名为水稻田 (Site 3, S3)、轮作农田 (Site 4, S4),轮种作物为:玉米-高粱-大豆;在保护区实验区设置2块退耕湿地 (CCTW),分别命名为水田退耕 (Site 5, S5)、旱田退耕 (Site 6, S6),样地详细信息如表1。
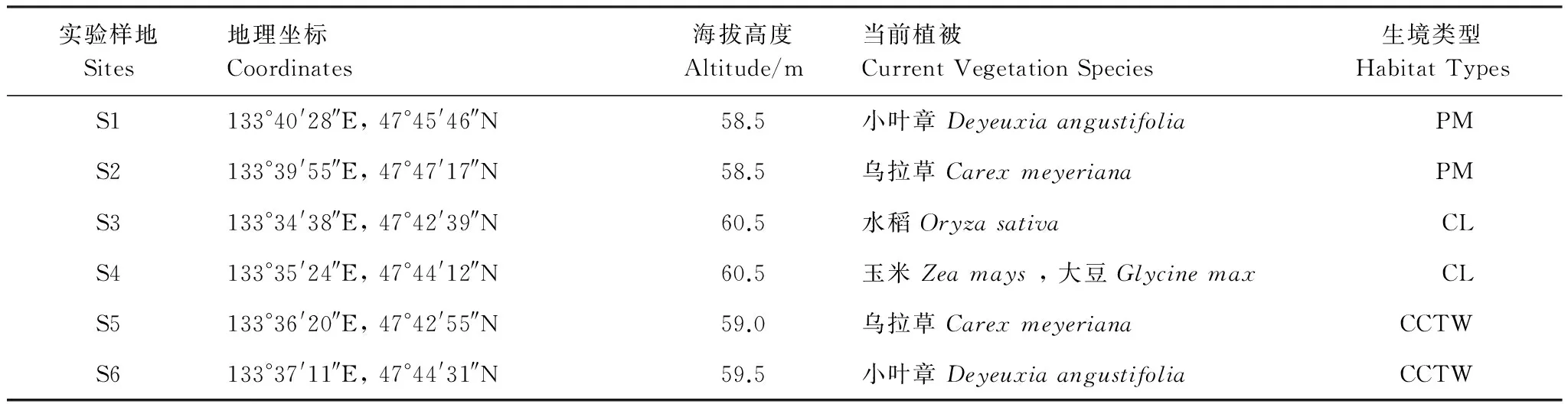
表1 样地的具体信息
PM: 原始沼泽 pristine marshland;CL: 耕地 cultivated land;CCTW: 退耕湿地 cropland converting to wetland
于2015年6月,土壤微生物量较大时进行土壤采集。每块实验样地内共设置了2块10 m×10 m的标准样方,在每个标准样方中随机布设2 m×2 m的小样方3个,分别在样方的四角与中心布设5个取样点,采用无菌土钻和环刀取表层大约5—20 cm深度的土壤样品,更换样地取样前对土钻等工具采取清洗杀菌措施。每个取样点中大约取出0.2 kg的土壤样品,然后将每个土样分为2份,一份土样装入无菌自封样品袋中并置于4℃电子恒温箱中,6块样地中共获取180份土样,用于测定土壤化学性质。将同一标准样方的剩余15份土样充分混合,过2 mm筛网,去除杂草与石块后装入无菌自封样品袋中,并储存于-80℃的干冰保温箱中,用于提取土壤细菌DNA,共获取12个土壤微生物样品[20]。
1.3 土壤理化指标的测定
土壤pH值用土∶水=1∶2.5浸提,电位法测定;土壤水分含量(Mc)使用重量分析仪测得;土壤的全碳(TC)和全氮(TN)含量使用元素分析仪测得;土壤有机碳(SOC) 使用岛津TOC-VCPH分析仪测定;土壤溶解有机碳(DOC)含量使用高纯度水浸提,取上清液过0.45m滤膜后使用岛津TOC-VCPH分析仪测定;碱解氮(AN)含量采用碱解蒸馏法测定;土壤微生物生物量碳(MBC)含量采用氯仿熏蒸-K2SO4浸提法,通过公式:MBC =EC/0.45计算得出,式中,EC为熏蒸和未熏蒸样品浸提液测定的有机碳差值;土壤微生物生物量氮(Soil microbial biomass of nitrogen, MBN)含量采用氯仿熏蒸-K2SO4浸提法,通过公式:MBN =EN/0.54计算得出,式中,EN为熏蒸和未熏蒸样品浸提液测定的有机氮差值[21- 23]。
1.4 土壤细菌DNA提取与PCR扩增
使用E.Z.N.A.®Soil DNA试剂盒(Omega U.S.)并按照说明书抽提基因组DNA,利用1%琼脂糖凝胶电泳检测抽提的基因组DNA,然后进行PCR扩增,在土壤细菌16SRNA的v4—v5区域合成带有条形码的特异引物。
引物设计:上游引物338F(5′- ACTCCTACGGGAGGCAGCA- 3′);
下游引物806R(5′- GGACTACHVGGGTWTCTAAT- 3′)[24]。
PCR试验采用TransGen AP221-02:TransStart Fastpfu DNA Polymerase,20L反应体系:5×FastPfu Buffer 4L, 2.5 mmol/L dNTPs 2L, Forward Primer(5mol/L) 0.8L, Reverse Primer(5mol/L) 0.8L, FastPfu Polymerase 0.4L, Template DNA 10ng, 补ddH2O至20L;PCR仪:ABI GeneAmp® 9700 型;
PCR反应参数:1) 1× (3 minutes at 95℃). 2) 27× (30 seconds at 95℃;30 seconds at 55 ℃;45 seconds at 72 ℃). 3) 10 minutes at 72 ℃, 10 ℃ until halted by user。全部样本严格按照正式实验条件进行,每个样本3个重复,将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测,使用AxyPrep DNA凝胶回收试剂盒(Axygen公司)切胶回收PCR产物[25- 26]。
1.5 Illumina Miseq测序
Tris_HCl洗脱,2%琼脂糖电泳检测,参照电泳初步定量结果,将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega 公司)进行检测定量。将纯化扩增产物等摩尔混合,构建Miseq文库,按照Illumina MiSeq平台的标准协议进行双端测序(2×300)。原始测序数据存放在美国国家生物技术信息中心建立了大容量数据库SRA中(http://www.ncbi.nlm.nih.gov/Traces/sra)。
1.6 生物信息统计与分析
Miseq测序序列数据首先根据 PE reads之间的 overlap关系,使用软件Trimmomatic将成对的 reads 拼接(merge)成一条序列,同时对 reads的质量和 merge的效果进行质控过滤,并根据序列首尾两端的barcode和引物序列区分样品得到有效序列,并校正序列方向。然后按照 barcode标签序列识别并区分样本得到有效数据。
数据去杂方法和参数:过滤read尾部质量值20以下的碱基,设置50bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,过滤质控后50bp以下的read;根据PE reads之间的overlap关系,将成对reads拼接(merge)成一条序列,最小overlap长度为10bp;拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列;根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2[26- 27]。
全部分析在运算分类单位 (Operational Taxonomic Units, OTU) 的相似水平为97% (0.97)条件下进行。使用Usearch软件 (version 7.1) 对所有序列进行OTU划分及生物信息统计;采用贝叶斯算法对OTU代表序列进行分类学分析,置信度阈值为0.7,比对RDP (Ribosomal Database Project)数据库 (Release 11.1);计算alpha多样性指数和做rarefaction分析采用mothur软件 (version v.1.30.1),计算公式:Ace指数参考 (http:// www.mothur.org/wiki/Ace);Chao指数 (http://www.mothur.org/wiki/Chao);Shannon指数 (http://www.mothur. org/wiki/Shannon);Simpson指数 (http://www.mothur.org/wiki/ Simpson)。利用R语言工具制作曲线图和文氏图;使用Qiime计算beta多样性距离矩阵,根据beta多样性距离矩阵进行层次聚类 (Hierarchical cluatering) 分析,使用非加权组平均法(Unweighted pair group method with arithmetic mean, UPGMA) 算法构建树状结构;R语言vegan包中冗余分析 (Redundancy Analysis, RDA) 和作图;使用SPSS软件 (IBM SPSS Statistics 22 for WINDOWS) 进行数据统计分析;差异显著性检验采用Duncan法和排列检验 (Permutation tests);相关性分析采用Pearson法。
2 结果与分析
2.1 不同生境类型样地土壤理化性质差异
垦殖及自然恢复对土壤pH值、含水量及碳、氮含量的影响差异达显著水平 (表2)。通过比较发现,耕地土壤的平均pH值比沼泽湿地土壤的高8.45%,比退耕湿地土壤的pH值高2.48%;耕地土壤含水量比沼泽湿地土壤的低41.37%,比退耕湿地土壤的含水量低13.05%;沼泽湿地土壤TC、TN、C/N、SOC、DOC、AN、MBC、MBN的含量分别是耕地土壤的2.36、1.19、1.97、2.38、3.23、1.43、4.42、3.18倍,分别是退耕湿地土壤的1.75、1.11、1.58、1.78、3.04、1.39、2.44、1.57倍。由此可见,持续的在沼泽湿地进行垦殖对土壤理化性质和微生物量具有显著的影响。
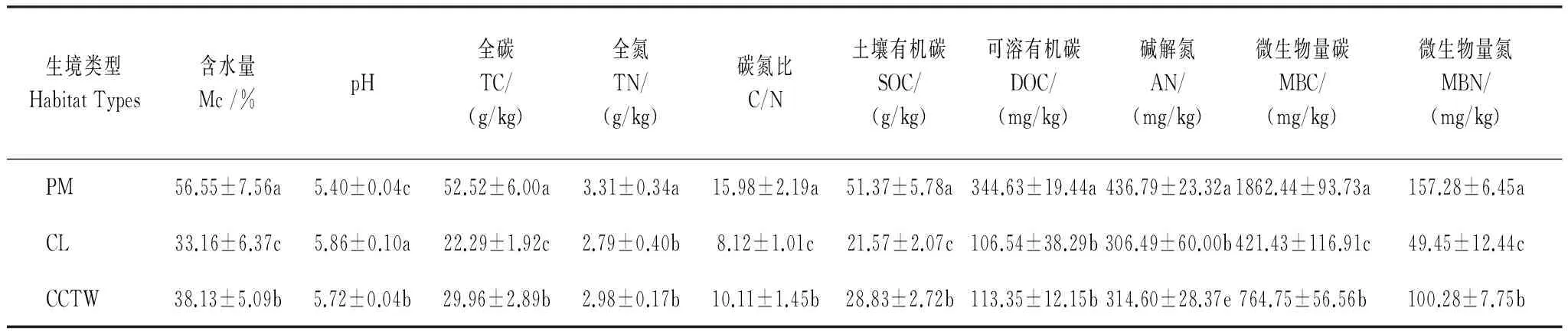
表2 不同样地的土壤理化性质和微生物量
不同小写字母表示不同生境类型间差异显著(P<0.05),最大的平均数标记为a,表中数据为平均值±标准差(n=180)
2.2 高通量测序数据分析
通过 miseq高通量测序并优化后,6个样地的12个土壤样品共获得358737条修剪序列且被划分为36个已知的菌门,总碱基数为1567500132,平均碱基长度为 436.95 bp,其中 401—500 bp的碱基占总序列数的99.89%。
稀释性曲线是从样本中随机抽取一定数量的个体,统计这些个体所代表的物种数目,并以个体数与物种数来构建曲线[28]。采用抽样序列数与它们所代表的OTU数目构建稀释性曲线(图1),可以看出12个土壤样品的稀释性曲线均趋于平坦,表明测序数据合理,更多的测序数据只会产生少量新的OTU。
2.3 不同类型样地土壤细菌群落的OTU分布
Venn图用于统计多个样本中所共有和独有的OTU数目,可以比较直观的表现环境样本的OTU数目组成相似性及重叠情况[29]。如图2,在97%的相似水平上进行统计,全部样地的土壤细菌OTU分布数目为2263个,其中共有的OTU分布数目为1411个,约占62.35%。退耕湿地土壤独有的土壤细菌OTU分布数目最少,约占全部的1.55%;耕地土壤独有的土壤细菌OTU分布数目最多,约占全部9.63%;原始沼泽湿地土壤独有的土壤细菌OTU分布数目为52个,约占全部的2.30%。原始沼泽湿地土壤与耕地土壤共有的土壤细菌OTU分布数目约占二者全部的67.77%;原始沼泽湿地土壤与退耕湿地土壤共有的土壤细菌OTU分布数目约占二者全部的75.21%;耕地土壤与退耕湿地土壤共有OTU分布数目约占全部的78.38%。由此可以看出原始沼泽湿地与耕地土壤细菌的OTU分布差异最显著。
2.4 不同类型土壤细菌群落α多样性
通过对各土壤样品细菌群落进行α多样性统计(表3),并对3种生境类型样地土壤细菌群落α多样性指数进行差异显著性检验可知,原始沼泽湿地土壤细菌群落的多样性相对较低,而耕作导致土壤细菌群落的多样性显著提高,湿地退耕后土壤细菌群落的多样性指数略有降低,与耕地土壤细菌群落的多样性差异水平不显著。
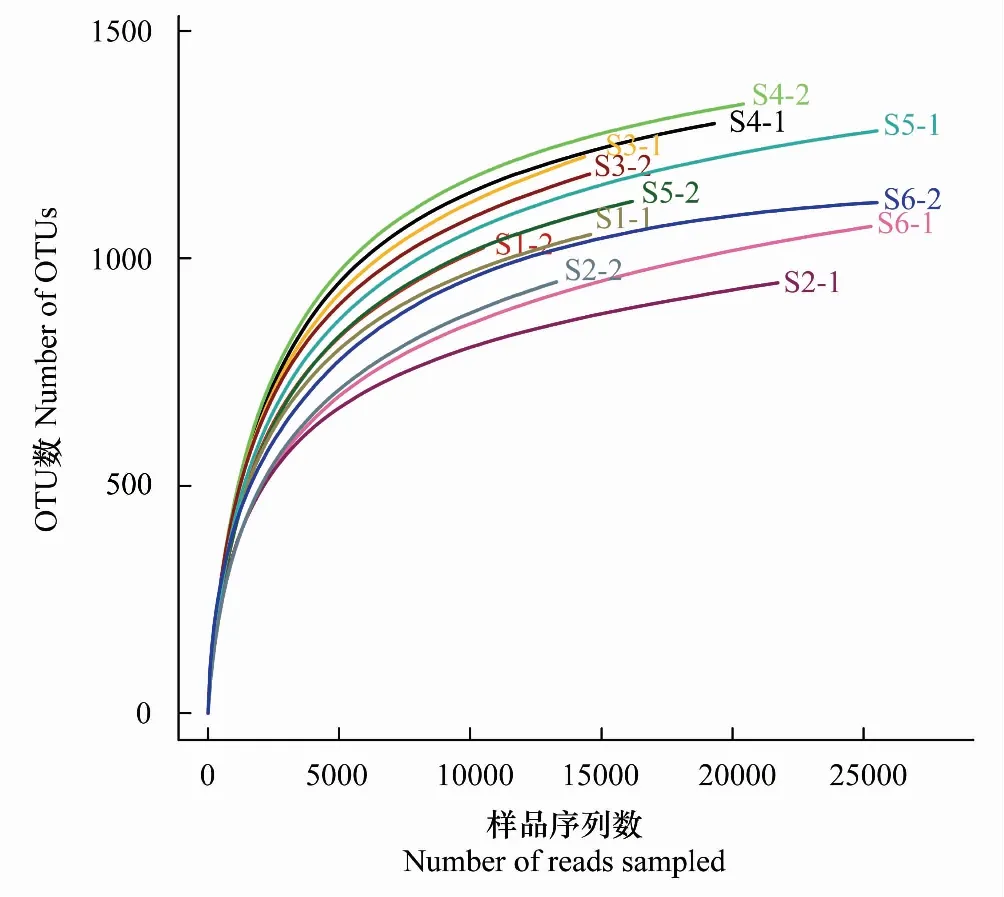
图1 不同样地土壤细菌OTU稀释曲线Fig.1 Rarefaction curves showing the extent of OTU detection at the different quadrats不同颜色的曲线代表不同样方;相似水平为97%;S:实验样地
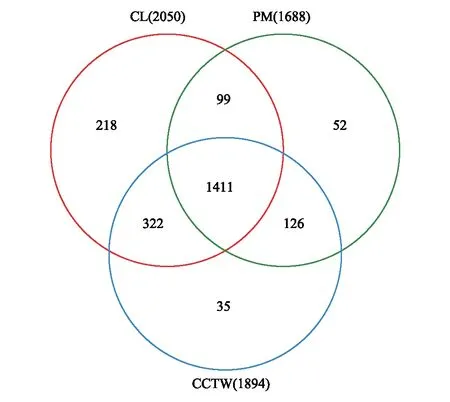
图2 不同类型样地土壤细菌基因测序获取的基因库中独有和共有OTUs分布的Venn图Fig.2 Venn diagram showing the numbers of shared and exclusive OTUs of soil bacteria identified at different types of sites PM, 原始沼泽pristine marshland;CL, 耕地cultivated land;CCTW, 退耕湿地cropland converting to wetland;相似水平:97%
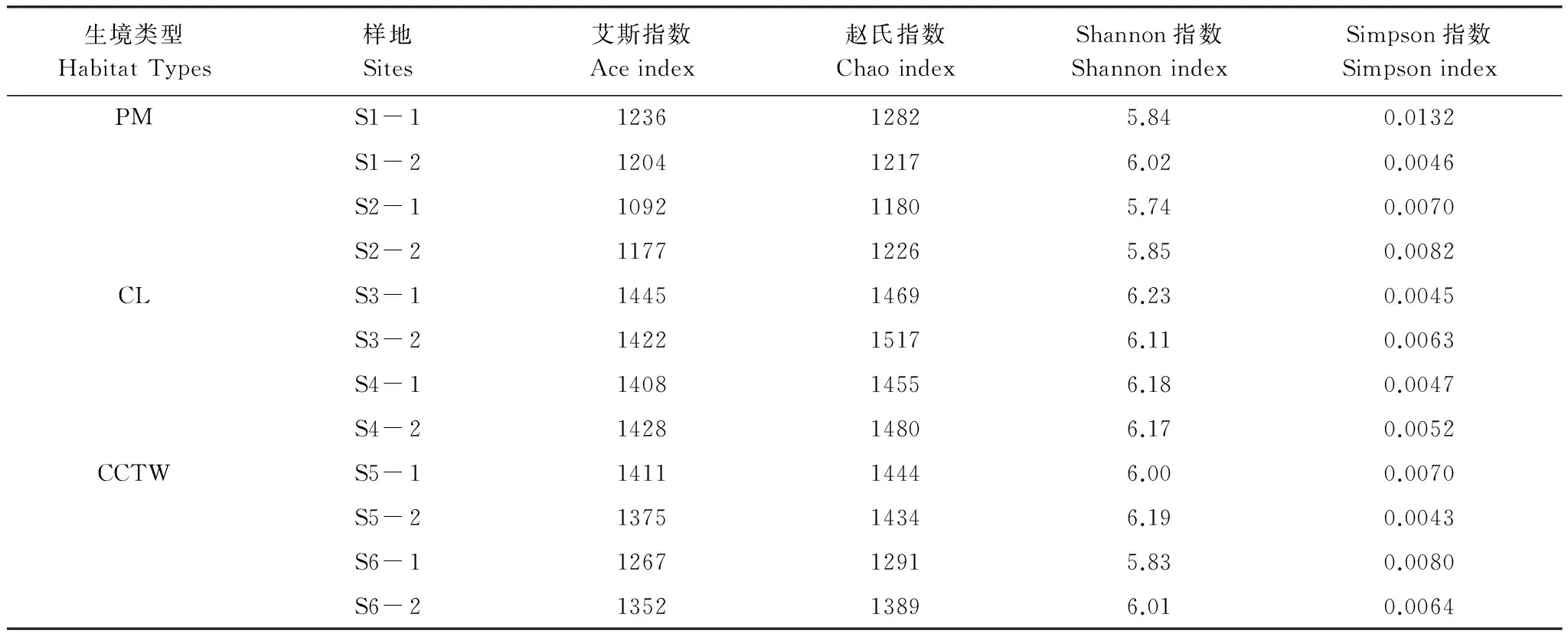
生境类型HabitatTypes样地Sites艾斯指数Aceindex赵氏指数ChaoindexShannon指数ShannonindexSimpson指数SimpsonindexPMS1-1123612825.840.0132S1-2120412176.020.0046S2-1109211805.740.0070S2-2117712265.850.0082CLS3-1144514696.230.0045S3-2142215176.110.0063S4-1140814556.180.0047S4-2142814806.170.0052CCTWS5-1141114446.000.0070S5-2137514346.190.0043S6-1126712915.830.0080S6-2135213896.010.0064
2.5 不同样地土壤细菌群落的分类组成及差异
在相似水平为 97%的条件下,对OTU的代表序列作分类学分析 (图3)。结果表明,12个土壤样品共检测出2263个OTUs分布,分属于36个菌门。其中,变形菌门 (Proteobacteria)、酸杆菌门 (Acidobacteria)、绿弯菌门 (Chloroflexi)、放线菌门 (Actinobacteria)、厚壁菌门 (Firmicutes)、芽单胞菌门(Gemmatimonadetes)、拟杆菌门 (Bacteroidetes)、疣微菌门 (Verrucomicrobia)、硝化螺旋菌门 (Nitrospirae)、(Saccharibacteria)和绿菌门 (Chlorobi)为主要细菌群落门类 (相对丰度>1%)。根据beta多样性距离矩阵进行层次聚类分析,使用非加权组平均法算法构建树状结构比较各样地土壤细菌群落的相似性和差异关系[30]。可以看出,原始沼泽湿地土壤菌群结构相似度较高,与耕地、退耕湿地间的差异较大,耕地与退耕样地之间的土壤菌群结构相似度较高。在原始沼泽湿地与耕地之间,豆田的菌群结构与乌拉草沼泽的菌群结构差异最高,而稻田的菌群结构与小叶章沼泽相似度。
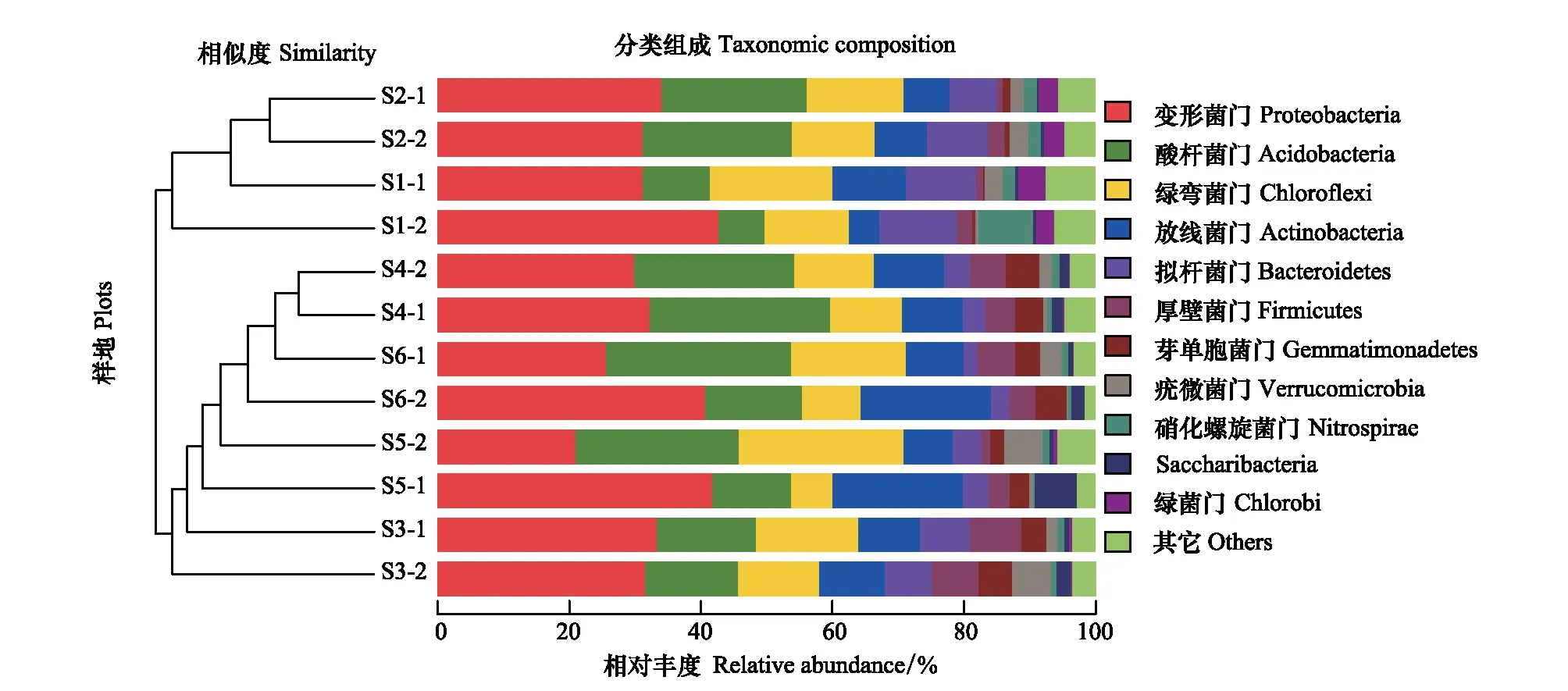
图3 不同样地土壤细菌门划分水平上的群落结构柱状图和聚类树Fig.3 Histogram and cluster tree of soil bacterial community in different plots
对不同生境类型样地11个主要细菌群落门类所占百分比采用Duncan法进行差异显著性检验。由此发现,变形菌门 (P=0.777)、酸杆菌门 (P=0.601)、绿弯菌门 (P=0.830)、放线菌门 (P=0.153)、疣微菌门 (P=0.950)、硝化螺旋菌门 (P=0.116)和Saccharibacteria (P=0.241)在各类型样地中分布较均匀,差异不显著;芽单胞菌门 (P<0.01)、拟杆菌门 (P<0.01) 和绿菌门 (P<0.01) 在无干扰样地与垦殖干扰样地、退耕湿地间差异显著;厚壁菌门 (P<0.01) 在3种类型样地间差异显著。
2.6 土壤细菌群落多样性与土壤性质、微生物量的关系
采用Pearson法分析计算样地土壤性质与细菌α多样性指数的相关性(表4),Ace指数、Chao指数、Shannon指数与含水量、TC、TN、C/N、SOC、DOC、AN、MBC、MBN显著负相关,与pH值显著正相关;Simpson指数与TC、C/N、SOC、DOC、AN、MBC、MBN显著正相关,与pH值显著负相关。
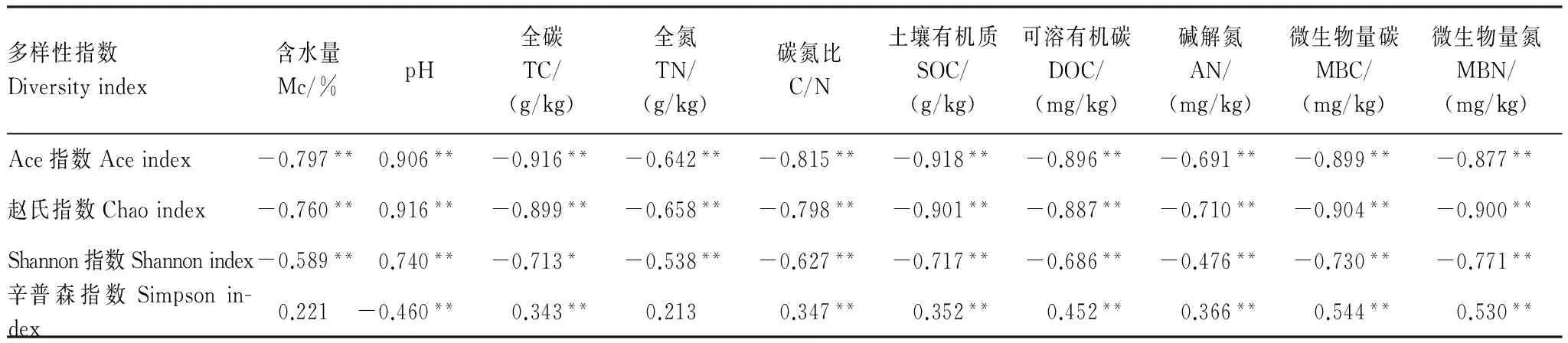
表4 土壤细菌α多样性指数与土壤理化性质、微生物量的相关系数
*P< 0.05;**P<0.01
对不同样地土壤细菌的 OTU (97%) 组成进行冗余分析,两轴累计贡献率为55.67% (图 4)。原始沼泽和水田退耕分布在第一排序轴正方向,耕地和旱田退耕样地分布在第一排序轴负方向,表明垦殖及自然恢复是第一排序轴的主要影响因素。Mc、TC、SOC、DOC和AN与第一排序轴正相关,TN和pH与第一排序轴负相关。排列检验显示 Mc、pH、TC、SOC和DOC对土壤细菌群落结构的影响最显著。
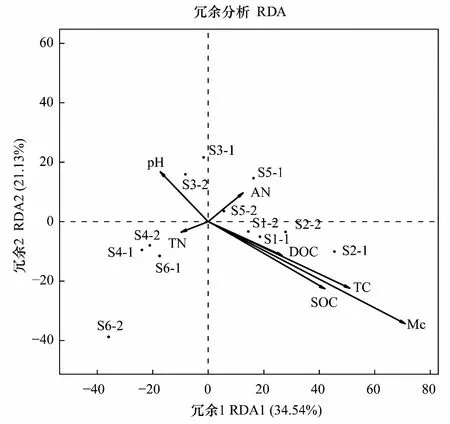
图4 环境因素对土壤细菌群落结构特征影响的冗余分析Fig.4 RDA of soil bacterial community and environmental factors
3 讨论与结论
3.1 不同生境土壤细菌群落结构的差异性及控制因子
目前,有关土壤细菌群落方面的研究内容较落后且严重不足,因而土壤细菌被比喻为“生物暗物质”[31],针对垦殖及恢复对土壤细菌群落影响机理等方面的科学问题亟待解决[1,18]。本研究采用新一代高通量测序技术对沼泽湿地土壤细菌群落进行测定。结果发现,不同类型生境的土壤细菌群落结构存在显著差异性,从聚类角度分析,退耕湿地与原始沼泽的土壤细菌群落结构差异较大而植被类型相同,表明植被类型对土壤细菌群落结构的影响处于次要地位。垦殖与退耕湿地的土壤细菌群落结构比较接近,与原始沼泽土壤细菌群落结构显著不同,可能是由于退耕还不能对土壤环境完全修复,有些相似的土壤性质导致土壤细菌存在相似种群[32]。
土壤细菌群落结构的变化以群落组分变化为基础,不同类型生境土壤细菌群落组分的不同预示着环境改变为这些细菌提供了特异性的生存条件,从而对菌群进行选择[33]。三江平原3种不同生境类型的沼泽湿地土壤细菌群落分属于36菌门,在垦殖和自然恢复的土样中没有检测出LCP- 89、TM6、Gracilibacteria、互养菌门 (Synergistetes) 的菌群,而在原始沼泽土样中没有检测出热袍菌门 (Thermotogae) 和梭杆菌门 (Fusobacteria) 的菌群,这说明垦殖过程破坏了原有土壤细菌群落的生态平衡,改变土壤菌群原有的生境条件,从而导致某种土壤细菌种群异常的增加或减少,同时引入了新的菌群为其提供适宜的生境条件,通常土壤细菌群落多样性会变得比原来高[34]。
不同类型生境的土壤样品中存在特有的高相对丰度和低相对丰度细菌门类[35]。如垦殖土样中的厚壁菌和芽单胞菌的相对丰度显著高于原始沼泽土样,而垦殖土样中的疣微菌和绿菌的相对丰度显著低于原始沼泽土样,并且这4种菌门的土壤细菌群落与土壤pH值、含水量及碳氮含量同时具有趋势性变化,表明这4种菌门的土壤细菌群落更易受垦殖干扰的影响,且土壤pH值、含水量及碳氮含量都是与沼泽土壤菌群变化密切相关的影响因子。沼泽土壤中含有庞大的细菌数量意味着拥有复杂的土壤细菌群落结构,认识和利用土壤细菌群落对维护土壤生态系统平衡以及促进耕地的可持续利用具有重要的意义[36]。
3.2 不同生境土壤细菌群落多样性的比较及控制因子
利用多样性指数分析土壤细菌群落是一种非常有效的方法[37],在研究土壤细菌α多样性时发现,垦殖导致土壤细菌群落多样性和丰富度显著高于原始沼泽,但湿地退耕并没有导致土壤细菌群落多样性和丰富度的显著降低,这与于振华利用DGGE图谱分析结果相似,在作物的生长季节耕地土壤细菌Shannon指数 (3.49—3.69) 高于自然恢复土地土壤细菌的Shannon指数(3.34—3.44)[38],海伦市与三江平原的纬度较为相近,耕地土壤条件具有相似性。刘方春等在研究持续干旱对土壤细菌群落多样性的影响中发现,随着持续干旱强度的增大,土壤细菌的数量及群落多样性先增加后减少[39],这与本研究旱田土壤细菌群落多样性高于水田和沼泽湿地的结果相似。三江平原夏季气候湿润,土壤含水量高,旱田耕作降低土壤含水量可以提高土壤细菌群落多样性,而干旱程度没有到达土壤细菌的群落多样性由增到减的阈值,沼泽湿地的土壤含水量可能是土壤细菌群落多样性的关键控制因子。
沼泽湿地垦殖或恢复的过程能够通过改变土壤的水热条件、土壤结构以及土壤营养条件,间接的影响土壤细菌群落结构和多样性[40]。土壤细菌群落多样性发生变化是适应垦殖干扰的直接表现,这种变化加快了土壤有机质的分解,也促进了作物吸收土壤养分[41]。本研究中发现,垦殖过程导致土壤中的TC、TN、SOC、DOC、AN、MBC及MBN含量显著降低,却显著提高了土壤细菌群落的多样性水平,推断出土壤碳、氮含量降低可以提高土壤细菌群落多样性,然而这种假设显然不合常理,这种现象有可能是因为原始沼泽土壤有机物经过常年积累,虽然土壤养分含量极高,但其自然存在形式不能够被大多数土壤细菌作为能量来源直接利用,垦殖改变了沼泽湿地土壤养分的原始存在形式,为土壤细菌对碳、氮的利用提供了有利条件,因此不能理解为土壤碳氮含量下降促进了土壤细菌多样性提高,而是只能说明沼泽湿地中的碳、氮含量并不是土壤细菌多样性的关键限制因子。本文研究了垦殖及自然恢复导致11个环境因子的变化对土壤细菌群落的影响,但仍缺少对土壤温度、土壤氧气含量等相关指标的测定,其影响机理有待进一步的研究。
致谢:黑龙江省洪河国家级自然保护区管理局提供帮助,哈尔滨工程大学刘迪博士和东北林业大学于洋博士在野外作业中给予技术支持,特此致谢。
[1] 王宗明, 宋开山, 刘殿伟, 张柏, 张树清, 李方, 任春颖, 金翠, 杨婷, 张春华. 1954—2005年三江平原沼泽湿地农田化过程研究. 湿地科学, 2009, 7(3): 208- 217.
[2] Liu J J, Sui Y Y, Yu Z H, Shi Y, Chu H Y, Jin J, Liu X B, Wang G H. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China. Soil Biology and Biochemistry, 2014, 70: 113- 122.
[3] 黄靖宇, 宋长春, 宋艳宇, 刘德燕, 万忠梅, 廖玉静. 湿地垦殖对土壤微生物量及土壤溶解有机碳、氮的影响. 环境科学, 2008, 29(5): 1380- 1387.
[4] Zhang X M, Zhang G M, Chen Q S, Han X G. Soil bacterial communities respond to climate changes in a temperate steppe. PLoS One. 2013, 8(11): e78616.
[5] 白雪, 马克明, 杨柳, 张洁瑜, 张小雷. 三江平原湿地保护区内外的生态功能差异. 生态学报, 2008, 28(2): 620- 626.
[6] 霍莉莉, 邹元春, 郭佳伟, 吕宪国. 垦殖对湿地土壤有机碳垂直分布及可溶性有机碳截留的影响. 环境科学, 2013, 34(1): 283- 287.
[7] 郭建国, 赵龙浩, 徐丹, 孙野青. 人工湿地处理造纸废水后细菌群落结构变化. 生态学报, 2014, 34(8): 2095- 2101.
[8] 张乃莉, 郭继勋, 王晓宇, 马克平. 土壤微生物对气候变暖和大气N沉降的响应. 植物生态学报, 2007, 31(2): 252- 261.
[9] 肖烨, 黄志刚, 武海涛, 吕宪国. 三江平原4种典型湿地土壤碳氮分布差异和微生物特征. 应用生态学报, 2014, 25(10): 2847- 2854.
[10] Ribeiro H, Mucha A P, Almeida C M R, Bordalo A A. Bacterial community response to petroleum contamination and nutrient addition in sediments from a temperate salt marsh. Science of the Total Environment, 2013, 458- 460: 568- 576.
[11] Sura S, Waiser M J, Tumber V, Raina-Fulton R, Cessna A J. Effects of a herbicide mixture on primary and bacterial productivity in four prairie wetlands with varying salinities: an enclosure approach. Science of the Total Environment, 2015, 512- 513: 526- 539.
[12] Arroyo P, de Miera L E S, Ansola G. Influence of environmental variables on the structure and composition of soil bacterial communities in natural and constructed wetlands. Science of the Total Environment, 2015, 506- 507: 380- 390.
[13] 汪仲琼, 王为东, 祝贵兵, 尹澄清. 人工和天然湿地芦苇根际土壤细菌群落结构多样性的比较. 生态学报, 2011, 31(16): 4489- 4498.
[14] Liu J J, Zheng C Y, Song C C, Guo S D, Liu X B, Wang G H. Conversion from natural wetlands to paddy field alters the composition of soil bacterial communities in Sanjiang Plain, Northeast China. Annals of Microbiology, 2014, 64(3): 1395- 1403.
[15] Ludwig-Müller J. Bacteria and fungi controlling plant growth by manipulating auxin: balance between development and defense. Journal of Plant Physiology, 2015, 172: 4- 12.
[16] 王兴春, 杨致荣, 王敏, 李玮, 李生才. 高通量测序技术及其应用. 中国生物工程杂志, 2012, 32(1): 109- 114.
[17] 刘驰, 李家宝, 芮俊鹏, 安家兴, 李香真. 16S rRNA基因在微生物生态学中的应用. 生态学报, 2015, 35(9): 2769- 2788.
[18] 袁红朝, 吴昊, 葛体达, 李科林, 吴金水, 王久荣. 长期施肥对稻田土壤细菌、古菌多样性和群落结构的影响. 应用生态学报, 2015, 26(6): 1807- 1813.
[19] 朱宝光. 三江原始湿地的“缩影”——黑龙江洪河国家级自然保护区. 生命世界, 2010, (5): 16- 19.
[20] 杨菁, 周国英, 田媛媛, 刘倩丽, 刘成锋, 杨权, 周洁尘. 降香黄檀不同混交林土壤细菌多样性差异分析. 生态学报, 2015, 35(24): 8117- 8217.
[21] 孙又宁, 保万魁, 余梅玲. 自动定氮仪碱解蒸馏法测定土壤中水解性氮含量. 中国土壤与肥料, 2007, (5): 64- 66.
[22] 鲁如坤. 土壤农业化学分析方法. 北京: 中国农业科技出版社, 2000: 125- 233.
[23] 李玉洁, 王慧, 赵建宁, 皇甫超河, 杨殿林. 耕作方式对农田土壤理化因子和生物学特性的影响. 应用生态学报, 2015, 26(3): 939- 948.
[24] Dennis K L, Wang Y W, Blatner N R, Wang S Y, Saadalla A, Trudeau E, Roers A, Weaver C T, Lee J J, Gilbert J A, Chang E B, Khazaie K. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL- 10-producing T cells. Cancer Research, 2013, 73(19): 5905- 5913.
[25] Schloss P D, Gevers D, Westcott S L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-Based studies. PLoS ONE, 2011, 6(12): e27310.
[26] 张芳, 林绍艳, 徐颖洁. 水稻连作对江苏地区稻田土细菌微生物多样性的影响. 山东农业大学学报: 自然科学版, 2014, 45(2): 161- 165.
[27] Yang S Z, Wen X, Jin H J, Wu Q B. Pyrosequencing investigation into the bacterial community in permafrost soils along the China-Russia Crude oil pipeline (CRCOP). PLoS One, 2012, 7(12): e52730.
[28] Amato K R, Yeoman C J, Kent A, Righini N, Carbonero F, Estrada A, Gaskins H R, Stumpf R M, Yildirim S, Torralba M, Gillis M, Wilson B A, Nelson K E, White B A, Leigh S R. Habitat degradation impacts black howler monkey (Alouattapigra) gastrointestinal microbiomes. The ISME Journal, 2013, 7(7): 1344- 1353.
[29] Fouts D E, Szpakowski S, Purushe J, Torralba M, Waterman R C, MacNeil M D, Alexander L J, Nelson K E. Next generation sequencing to define prokaryotic and fungal diversity in the Bovine Rumen. PLoS One, 2012, 7(11): e48289.
[30] Jiang X T, Peng X, Deng G H, Sheng H F, Wang Y, Zhou H W, Tam N F Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a Mangrove wetland. Microbial Ecology, 2013, 66(1): 96- 104.
[31] Jansson J K, Prosser J I. Microbiology: the life beneath our feet. Nature, 2013, 494(7435): 40- 41.
[32] Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiology Ecology, 2009, 68(1): 1- 13.
[33] 苗祯, 杜宗军, 李会荣, 楼妍颖, 罗玮. 5株北极微藻藻际环境的细菌多样性. 生态学报, 2015, 35(5): 1587- 1600.
[34] 陈波, 黄霄, 刘小玉, 周登博, 谭昕, 高祝芬, 张锡炎, 戚春林. 不同香蕉枯萎病区土壤细菌群落多样性. 应用生态学报, 2013, 24(8): 2281- 2286.
[35] 张伟, 陈一峰. 棉花长期连作对新疆土壤细菌群落结构的影响. 生态学报, 2014, 34(16): 4682- 4689.
[36] Cookson W R, Murphy D V, Roper M M. Characterizing the relationships between soil organic matter components and microbial function and composition along a tillage disturbance gradient. Soil Biology and Biochemistry, 2008, 40(3): 763- 777.
[37] 隋心, 张荣涛, 钟海秀, 许楠, 王继丰, 刘应竹, 袁海峰, 倪红伟. 利用高通量测序对三江平原小叶章湿地土壤细菌多样性的研究. 土壤, 2015, 47(5): 919- 925.
[38] 于镇华. 垦殖与自然恢复黑土微生物群落结构及生态功能的季节变化[D]. 北京: 中国科学院研究生院, 2012.
[39] 刘方春, 邢尚军, 马海林, 陈波, 丁延芹, 杜秉海. 持续干旱对樱桃根际土壤细菌数量及结构多样性影响. 生态学报, 2014, 34(3): 642- 649.
[40] Sheik C S, Mitchell T W, Rizvi F Z, Rehman Y, Faisal M, Hasnain S, McInerney M J, Krumholz L R. Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS One, 2012, 7(6): e40059.
[41] Zhang X M, Chen Q S, Han X G. Soil bacterial communities respond to mowing and nutrient addition in a steppe ecosystem. PLoS One, 2013, 8(12): e84210.
Effect of cultivation and natural restoration on soil bacterial community diversity in marshland in the Sanjiang Plain
XU Fei1, 2, CAI Tijiu1,*, YANG Xue1, 2, JU Cunyong1, TANG Qingming3
1CollegeofForestry,NortheastForestryUniversity,Harbin150040,China2HeilongjiangAcademyofLandReclamationSciences,Harbin150038,China3HulunbeierForestryAdministration,Hulunbeier021008,China
Different perturbation regimes, including disturbance caused by cultivation or the process of natural restoration, can have significant effects on the soil bacterial community in marshland. In this study, we investigated the relationship between soil bacterial community composition and perturbation in marshland to quantify the extent of such disturbance-related changes in northeast China. We assessed the diversity of bacterial communities in twelve samples of marsh soil collected from pristine marsh, neighboring cropland, and a wetland restoration area. High-throughput sequencing of a bacteria-specific genomic sequence, the internal transcribed spacer (16S rRNA) region, was used to identify bacterial taxa. We obtained 358,737 sequences that represented 2263 bacterial OTUs across the three types of sampling sites. Of these, 1411 OTUs occurred at all three site types, 99 were shared between cultivated land and pristine marshland, 322 were shared between cultivated land and wetland converted from cropland, and 126 were shared between pristine marshland and wetland converted from cropland. All sites also hosted unique fungal OTUs, with 218 OTUs exclusive to cultivated land, 52 exclusive to pristine marshland, and 35 exclusive to wetland converted from cropland. Sequences were affiliated to 36 different phyla throughout the dataset. Sequence abundance showed that members of the Proteobacteria were more frequently identified in all soil samples than Acidobacteria, and included members of Chloroflexi, Actinobacteria, Bacteroidetes, Firmicutes, Gemmatimonadetes, Verrucomicrobia, Nitrospirae, Saccharibacteria, and Chlorobi, which represented an overwhelming proportion of the soil bacterial communities with an average relative abundance of > 1%, and another 25 phyla with an average relative abundance were < 1%. The dominant phyla that showed the greatest variation among habitat types (> 1% of the average relative abundance) were Gemmatimonadetes (P<0.01), Bacteroidetes (P<0.01), Firmicutes (P<0.01), and Chlorobi (P<0.01). The soil bacterial community diversity decreased from a maximum in cultivated land, through the wetland restoration area, to a minimum in pristine marshland. Redundancy and correlation analyses demonstrated that chronic disturbance through cultivation, especially dry cultivation, significantly altered the bacterial community composition of marsh soil. The α-diversity of the soil bacterial community was most affected by soil moisture, soil pH, total carbon, soil organic carbon, soil dissolved organic carbon, available nitrogen, microbial biomass of carbon, and microbial biomass of nitrogen. Meanwhile, the soil bacterial community composition was significantly affected by soil moisture, soil pH, total carbon, soil organic carbon, and soil dissolved organic carbon. Overall, the results from our study showed that the state of soil carbon and nitrogen is affected by the disturbance by agricultural cultivation, causing long-term accumulated soil nutrients to become available as an energy source that can be rapidly mineralized by soil bacteria. In addition, our results also indicate that cultivation and natural restoration influenced the bacterial community structure and diversity. Natural restoration can significantly enhance the recovery of bacterial diversity; however, once the composition of the marshland bacterial community has been altered by cultivated disturbance, it might be difficult to restore to its original state. These findings highlight the importance of effectively managing the soil bacterial community to maintain a naturally functioning soil ecosystem.
marsh; cultivation; soil bacteria; α-diversity; community structure; high-throughput sequencing
黑龙江省青年科学基金项目(QC2014C029);国家水体污染控制与治理科技重大专项课题 (2015ZX07201-008-03)
2016- 01- 04;
2016- 07- 25
10.5846/stxb201601040015
*通讯作者Corresponding author.E-mail: caitj@263.net
徐飞, 蔡体久, 杨雪, 琚存勇, 唐庆明.三江平原沼泽湿地垦殖及自然恢复对土壤细菌群落多样性的影响.生态学报,2016,36(22):7412- 7421.
Xu F, Cai T J, Yang X, Ju C Y, Tang Q M.Effect of cultivation and natural restoration on soil bacterial community diversity in marshland in the Sanjiang Plain.Acta Ecologica Sinica,2016,36(22):7412- 7421.
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
现代园艺(2021年23期)2021-12-01
食品安全导刊(2021年20期)2021-08-30
林业勘查设计(2020年1期)2021-01-18
新农业(2020年18期)2021-01-07
下一代英才(酷炫少年)(2017年9期)2017-11-27
剑南文学(2016年11期)2016-08-22
水生生物学报(2015年1期)2015-02-28
时代英语·高三(2014年5期)2014-08-26
河南科技(2014年18期)2014-02-27
