
受体靶向多肽载体抗肿瘤药物
2016-01-21 03:11孙立春CoyDH美国杜兰大学医学院多肽药物研发中心
上海医药 2015年1期
孙立春Coy DH(美国杜兰大学医学院多肽药物研发中心)
受体靶向多肽载体抗肿瘤药物
孙立春*Coy DH
(美国杜兰大学医学院多肽药物研发中心)
摘 要常规化疗药物对癌细胞没有选择性,常常会导致严重的副作用。提高这些药物的靶向特异性已成为药物开发的热点方向之一。一些小分子多肽能够靶向作用于特定的受体,因而被用作癌症化疗药物的载体。化疗药物与多肽载体偶联构成新的多肽载体抗肿瘤药物。这些药物具有高特异性、高亲和力和肿瘤渗透力等优点,能够通过细胞表面的特定受体将药物送到靶向癌细胞内,提高抗癌效果、减少副作用和癌细胞的耐药性。多肽载体靶向药物被誉为新一代的靶向特异性的抗肿瘤药物之一。
关键词受体 多肽载体 肿瘤 靶向治疗 丙戊酸
Receptor-targeted cytotoxic peptide-drug conjugates
SUN Lichun*, David H. Coy
(Department of Medicine, Peptide Research Laboratories, Tulane University Health Sciences Center, New Orleans, LA 70112, USA)
ABSTRACTConventional cancer chemotherapy has very limited effects due to lacking specificity resulting in severe toxic side effects. Certain G protein-coupled receptors (GPCRs) are highly expressed in many tumor cells and tumoral blood veins, with their cognate ligands being peptides. Therefore, these peptides, especially their long-acting analogs, can be applied as drug-delivery vehicles by coupling with cytotoxic agents. These novel cytotoxic peptide-drug conjugates display more potent anti-tumor efficacy by targeting the cognate receptors while reducing toxic side effects and overcoming multiple drug resistance. This new receptor-targeted approach may provide a promising opportunity for the improvement of cancer treatments.
KEY WORDSreceptor-targeted; peptide-drug conjugate; tumor; VPA
目前治疗癌症最常用和最有效的方法就是通过手术来切除癌变组织,辅之以化疗、放疗。对不能手术的癌症患者或者癌症转移的晚期患者,放疗和化疗更是最有效的手段。然而,常规的化疗放疗药物对细胞没有选择性,不可避免对正常细胞的伤害。因此,开发針对癌症的特定靶向药物或提高化疗药物对癌细胞的靶向特异性,能够大大提高对癌症的治疗效果。100多年前,Ehrlich就提出魔术子弹(magic bullet)的概念[1]。这种魔术子弹药物能够特异性地识别和杀死癌细胞而不伤害正常细胞。美国杜兰大学医学院诺贝尔奖获得者Andrew V. Schally教授发明了用多肽作为癌症化疗药物的载体,将药物传递到受体特异性的靶向部位[2-3]。多肽作为靶向载体具有很多优越性和特异性,正吸引着越来越多的关注。
1 药物载体
就药物载体而言,研究得比较多的主要有单克隆抗体、纳米材料、多肽、脂质体和高分子等。其中,只有多肽和单克隆抗体作为载体具有高度的靶向特异性。单克隆抗体载体具有靶向特异性高、亲和力强、稳定性好等优点。缺点是仍然存在一些待解决的问题,如抗体为大分子,肿瘤渗透力差,载药量少,药物与抗体连接比例和连接效率的不确定性,抗原的异质性影响,抗体生产成本较高,批量生产比较复杂,网状内皮系统和肝脏对抗体的非特异性吸收[4-5]。而纳米材料的好处是纳米级小分子,对小分子或多肽药物的吸附力和载药量都很强,能将大量药物快速传送到组织细胞内。但是,纳米材料也很容易被正常组织细胞吸附而难以避免副作用,同时,不清楚纳米材料对人体的毒性和在人体内的稳定性,另外,如何提高药物靶向特异性等也是问题之一。至于脂
质体和高分子材料等作为药物载体更存在最基本的靶向性和特异性问题。
多肽作为载体的好处是小分子。人体内存在很多自然多肽,这些多肽容易被代谢和从体内清除,无明显副作用。多肽一般没有免疫原性,也不能穿过血脑屏障。天然多肽最大的问题是半衰期时间很短,但是通过改造修饰后会较稳定,这些经过修饰和改造后的长效多肽能够用作有效的药物载体。多肽作为靶向载体还拥有高亲和力、高靶向特异性和高稳定性等特点,而且生产工艺简单,容易工业化。多肽载体不仅能很快将所携带药物传递到特定细胞内,而且具有肿瘤渗透力。除了作为化疗药物载体外,多肽也可以广泛应用到其它方面,能够与各种其它药物分子相连,能够与siRNA、oligoDNA、oligoPNA (peptide nucleic acid)相连,也能够与单克隆抗体或者其它多肽相连,又能够与纳米材料、脂质体、多聚高分子结合,增强靶向特异性和细胞吸收[4-7]。
2 多肽载体靶向技术
多肽作为载体最初被应用于与放射性同位素偶联,用来进行放射治疗(radiotherapy)和造影(imaging)。应用于化疗药物的多肽载体靶向技术是由美国杜兰大学医学院教授、诺贝尔奖获得者Andrew V. Schally教授创造发明。他于1989年首次将小分子抗肿瘤药物溶肉瘤素(美法仑,melphalan)和苯丁酸氮芥(chlorambucil)分别与多肽促黄体激素释放激素(luteinizing hormone-releasing hormone,LHRH)连接,构成新的LHRH受体靶向抗肿瘤复合物[2-3]。LHRH受体属于G蛋白偶联受体(G protein-coupled receptor,GPCR)家族成员(GPCR是有近1 000个成员的庞大家族,为细胞表面受体)(图1)。自此以来,多肽作为受体靶向的抗肿瘤载体被越来越多地应用于改善化疗药物的效果,也被实验证明能够通过增强药物的靶向特异性,增强药物的靶向吸收,提高抗肿瘤效果,同时还能够改善一些小分子药物原有的难溶性等缺点。多肽载体靶向技术被誉为新一代的靶向药物开发技术[4-5,8-9]。
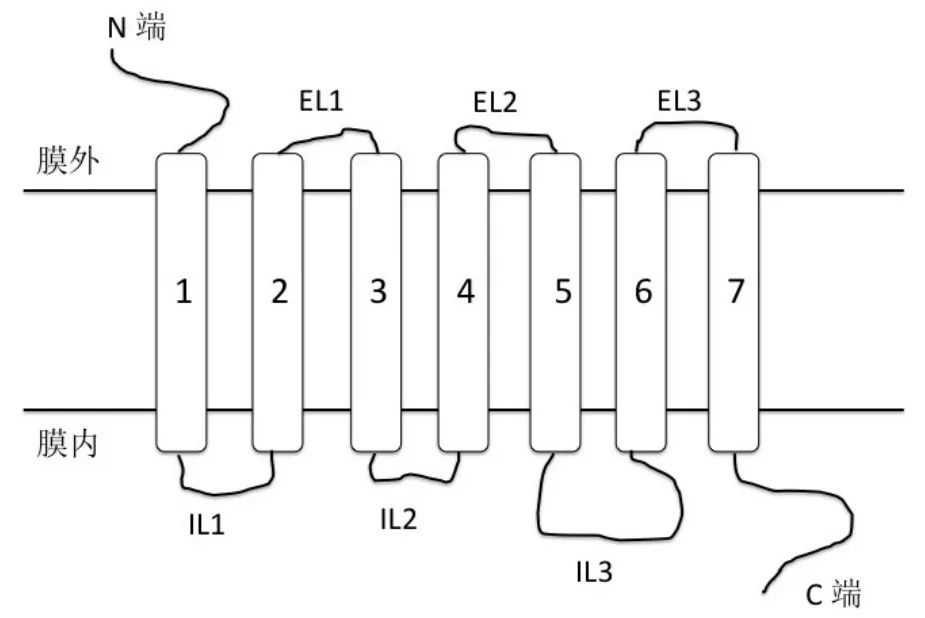
图1 G蛋白质偶联受体(GPCR)结构示意图(其主要特征是有7个跨膜结构域)
具体来说,多肽载体靶向技术是利用多肽作为载体,将化疗小分子药物与多肽载体相连,主要是与其N端或C端相连,构成新的受体靶向的多肽载体抗肿瘤复合物。这主要包括三部分:多肽载体、化疗药物和两者之间的连接链(linker,spacer)(图2)。因此,需要考虑三个主要方面:①对小分子药物进行筛选,了解其抗癌性能和分子结构上可能的连接点,或者对这些小分子药物进行改造或修饰,以利于连接,同时保持或提高小分子药物的活性。②筛选可能用作载体的多肽。天然多肽由于半衰期很短,一般不适于作为药物载体,需要对之进行改造或修饰,使之更加稳定,同时要保持或提高受体亲合力和受体特异性。出于对合成效率、生产成本等因素的考量,多肽载体分子的大小也需要考虑。多肽的空间结构也可能妨碍与化疗药物的有效连接,如,环状多肽比线型多肽有更好的稳定性和选择性。③寻找特定的连接方式将多肽载体与小分子化合物进行偶联,构成新的多肽载体抗肿瘤复合物(图2)。这种新的多肽载体抗肿瘤复合物既保持原多肽载体的特性,又确保这种复合物具有一定的稳定性,能将小分子药物有效地传递到特定癌细胞部位,并高效地释放这些小分子,达到更有效的抗肿瘤效果(图3)[8,10-11]。
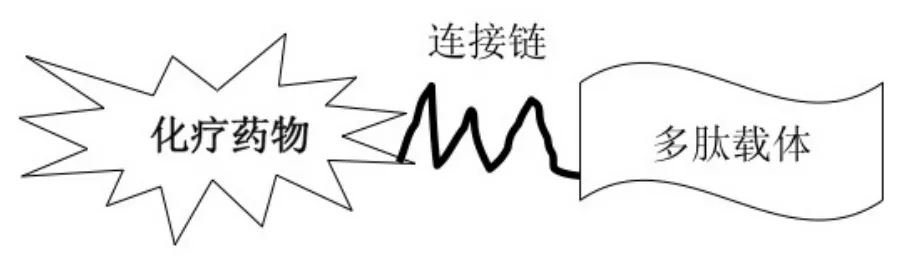
图2 受体靶向的多肽载体抗肿瘤药物结构示意图
3 多肽作为受体靶向的抗肿瘤药物载体的主要依据
由于小分子药物强大的抗癌效果和极其明显的缺陷,特别是长期使用引起的严重的不良反应和癌细胞产生的耐药性,直接导致人们用各种方法来改善或提高化疗效果。现已发现一些受体、蛋白或者酶在特定肿瘤细胞或者新生肿瘤血管中异常地高表达,而在正常组织器官中不表达或者表达量很低,利用这些表达差异来把药物导向靶向癌细胞就成为可能[10,13-19]。我们这里主要讨论GPCR受体靶向性的多肽载体抗肿瘤药物(图1,
2)。其中相当一部分GPCR的配体为多肽,如生长抑素(somatostatin,SST),促黄体激素释放激素(LHRH),胃泌素释放肽(gastrin-releasing peptide,GRP),垂体腺苷环化酶激活多肽(pituitary adenylate cyclase-activating polypeptide,PACAP),尾加压素(硬骨鱼紧张肽, urotensin II,UII)(表1)。与这些多肽相应的特异性受体在许多癌细胞中或者肿瘤血管中高水平地表达[11,16],使得这些多肽可作为抗肿瘤化疗药物的载体将药物通过特定受体更有效地传递到特定靶向肿瘤细胞,达到提高抗癌效果、减少副作用的目的[4-5,20]。

表1 一些多肽及其氨基酸序列
另外,小分子化疗药物由于分子量小,能通过细胞膜上的离子通道或以扩散的方式进入细胞,既能进入正常细胞,也能进入癌细胞,没有选择性。而癌细胞内的多药耐药基因(multiple drug-resistant gene)也可以将这些小分子药物外排到(pump out)至胞外,导致癌细胞对这些药物的耐药性。多肽载体抗肿瘤药物是通过细胞表面特定的GPCR受体将这些小分子药物传递到细胞内。这些复合物比小分子药物本身大很多,也比天然多肽更稳定,不会被很快降解,使得进入到细胞内的小分子药物不易被耐药基因直接外排出细胞。一些实验证明多肽载体还能够把药物进一步送到细胞核内,避免耐药基因的作用[5,7,20-23]。
4 受体靶向多肽载体抗肿瘤药物
自从Schally教授发明多肽载体靶向技术以来,各种化疗药物被尝试与各种多肽载体连接,构成各种受体靶向的多肽载体复合物,如生长抑素(SST)复合物(如JF-10-81、AN-162、AN-238)、促黄体激素释放激素(LHRH)复合物(如AN-152、AN-207)[24-26]、胃泌素释放肽(GRP)复合物[27-28]、垂体腺苷环化酶激活多肽(PACAP)复合物和尾加压素(UII)复合物等等。这些新的靶向复合物也展示了更好的靶向特异性和更有效的抗肿瘤效果[20-21,27,29]。生长抑素复合物JF-10-81是由长效生长抑素衍生物和抗癌药物喜树碱(camptothecin,CPT)构成的生长抑素Ⅱ型受体(SSTR2)靶向的复合物CPT-SST。在小鼠实验模型中,JF-10-81比抗癌药物CPT本身更有效地抑制胰腺癌(图3)、白血病、类
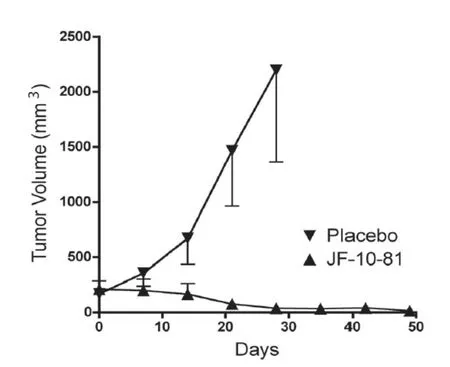
图3 JF-10-81(CPT-SST)抑制胰腺癌肿瘤生长(给药方式为不间断的缓释剂片。将含5 mg药物的片剂
植于小鼠皮下, 可以不间断地持续 60 d释放药物[12])
癌、神经母细胞瘤、前列腺癌和宫颈癌等多种肿瘤的生长[12,30-31]。促黄体激素释放激素(LHRH)复合物AN-152和AN-207是分别由2-吡咯霉素AN-201和阿霉素(doxorubicin)与LHRH载体偶联而成。这些复合物在治疗一系列的肿瘤如前列腺癌、乳腺癌、卵巢癌、非霍奇金淋巴瘤、子宫内膜癌、膀胱癌、肾细胞癌、恶性黑色素瘤、肝癌、胰腺癌、结肠直肠癌等都展示了比单独的化疗药物本身有更好的抗肿瘤效果[4,21,24-26,29]。
这些新的受体靶向的多肽载体抗肿瘤药物不仅能抑制肿瘤生长,还能抑制肿瘤血管生成(angiogenesis),克服癌细胞的耐药性。一个典型例子就是通过抑制肿瘤血管生成来抑制肿瘤生长。人非小细胞肺癌(non-SCLC)H-157细胞本身不表达人hSSTR2。而SSTR2受体靶向的多肽载体抗肿瘤药物AN-238能有效抑制小鼠皮下的人non-SCLC肿瘤的生长。Schally教授进一步发现,虽然H-157细胞不表达人hSSTR2,但是在小鼠皮下肿瘤血管中检测到丰富的小鼠mSSTR2,表明AN-238通过作用于小鼠mSSTR2,抑制小鼠皮下肿瘤血管生成来抑制人non-SCLC肿瘤生长,证明AN-238具有高度的SSTR2靶向特异性[32]。这些受体靶向复合物还能够抵御癌细胞的耐药性。如类癌BON细胞表达丰富的耐药基因产物MDR1和MRP1,能够抵御CPT的抗癌作用,但是,BON细胞也表达丰富的生长抑素受体(SSTR),多肽载体药物JF-10-81通过SSTR2将药物聚集到细胞内,非常明显地增强CPT对类癌BON肿瘤的抗肿瘤效果[8]。此外,AN-215对乳腺癌和肾细胞癌[22,33],AN-238对子宫内膜癌和黑色素瘤[23-34],AN-152对乳腺癌、卵巢癌、子宫内膜癌[35-36]都能克服耐药基因的影响,展现强抗癌效果。
5 多肽载体抗肿瘤药物与联合治疗
为提高癌症化疗效果,减少对特定器官产生的副作用和癌细胞的耐药性,多个化疗药物联合使用已经成为常规策略[37-38]。但是,这些药物本身没有选择性,联合治疗也不能完全避免对正常细胞的伤害。我们发现一些小分子化合物本身不仅有抗癌作用,而且能作为受体激活因子进一步增强特定受体的表达[37,39-40]。受体的增加能够更快更有效地促进药物的细胞吸收。这一特点为我们提供了一种全新的联合治疗的机会,将这种小分子化合物与其激活的特定受体的靶向多肽载体抗肿瘤药物联合应用,能更有效地增强抗肿瘤效果。如已发现丙戊酸(valproic acid,VPA;已经被美国FDA批准,临床应用于治疗癫痫患者)能够通过调控组蛋白去乙酰化酶(histone deacetylase,HDAC)和Notch信号来抑制肿瘤生长。VPA具有副作用小的特点,已经被广泛应用于各种癌症的联合治疗[37-38]。同时,我们发现VPA在一些癌细胞中能够激活一些GPCR受体的表达。例如VPA在肺癌、肝癌、类癌、卵巢癌和宫颈癌等癌细胞中能够抑制癌细胞生长,还能激活SSTR2的表达[23,34-36,39-40]。利用这一特点,可以预期VPA与SSTR2靶向的生长抑素载体抗肿瘤药物如JF-10-81(CPT-SST),COL-SST,AN-238,AN-152[26-27]联合使用,能够更有效地增强彼此的抗肿瘤效果。动物实验也进一步证明,VPA与JF-10-81 (CPT-SST)或VPA与COL-SST联合用药,比单独的VPA或JF-10-81(CPT-SST)或COL-SST更有效地抑制宫颈癌肿瘤生长(图4)。而且在联合用药的用药量大幅减少的情况下,既提高受体靶向的抗癌效果,又减少副作用[39-40]。我们在其它肿瘤的动物实验中也获得了类似结果(未发表)。这种联合治疗为癌症手术后或者晚期癌症的化疗提供了一种新的方式。

图4 VPA与COL-SST联合治疗在用药剂量减少的情况下能有效地增强抗肿瘤效果
6 展望
传统的化疗对很多癌症尤其晚期癌症作用有限。提高癌症化疗效果仍然是为人们所迫切期待的事。用长效多肽作为药物载体来提高癌症化疗的靶向性和特异性是我们值得努力的方向之一。这种新一代的受体靶向抗肿瘤复合物也展示了其良好的抗肿瘤效果。目前已有多个此类药物在各级临床试验中[4-5]。世界上第一个多肽靶向药物有望在未来几年内上市。
参考文献
[1] Ehrlich P. The relationship existing between chemical constitution, distribution, and pharmacological action[M]// Himmelweite F. The Collected Papers of Paul Ehrlich, Vol.1. Histology, Biochemistry and Pathology. New York: Pergamon Press, 1956: 596-618.
[2] Bajusz S, Janaky T, Csernus VJ, et al. Highly potent metallopeptide analogues of luteinizing hormone-releasing hormone[J]. Proc Natl Acad Sci USA, 1989, 86(16): 6313-6317.
[3] Bajusz S, Janaky T, Csernus VJ, et al. Highly potent analogues of luteinizing hormone-releasing hormone containing D-phenylalanine nitrogen mustard in position 6[J]. Proc Natl Acad Sci USA, 1989, 86(16): 6318-6322.
[4] Schally AV, Engel JB, Emons G, et al. Use of analogs of peptide hormones conjugated to cytotoxic radicals for chemotherapy targeted to receptors on tumors[J]. Curr Drug Deliv, 2011, 8(1): 11-25.
[5] Sun LC, Coy DH. Cytotoxic conjugates of peptide hormones for cancer chemotherapy[J]. Drugs Fut, 2008, 33(3): 217-223.
[6] Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy[J]. Endocr Rev, 2003, 24(4): 389-427.
[7] Schally AV, Engel JB, Emons G, et al. Use of analogs of peptide hormones conjugated to cytotoxic radicals for chemotherapy targeted to receptors on tumors[J]. Curr Drug Deliv, 2011, 8(1): 11-25.
[8] Sun L, Morris LM, Luo J, et al. Application of human pancreatic carcinoid BON cells for receptor-targeted drug development[J]. J Drug Target, 2011, 19(8): 666-674.
[9] Ahrens VM, Bellmann-Sickert K, Beck-Sickinger AG. Peptides and peptide conjugates: therapeutics on the upward path[J]. Future Med Chem, 2012, 4(12): 1567-1586.
[10] Forner B, Guiles J. Peptide-drug conjugates: Types, utility & manufacturing[J/OL]. Speciality Chemicals Magazine, 2012, 32: 46-47 [2014-01-13]. http://cedarburghauserpharma.com/ wp-content/uploads/peptide-drug-conjugates-specchem1.pdf.
[11] Sun L, Luo J, Mackey LV, et al. Investigation of cancer cell lines for peptide receptor-targeted drug development[J]. J Drug Target, 2011,19(8): 719-730.
[12] Sun LC, Mackey LV, Luo J, et al. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice[J]. Clin Med Oncol, 2008, 2: 491-499.
[13] Tai W, Shukla RS, Qin B, et al. Development of a peptidedrug conjugate for prostate cancer therapy[J]. Mol Pharm, 2011, 8(3): 901-912.
[14] Yamada R, Kostova MB, Anchoori RK, et al. Biological evaluation of paclitaxel-peptide conjugates as a model for MMP2-targeted drug delivery[J]. Cancer Biol Ther, 2010, 9(3): 192-203.
[15] Lee GY, Park K, Kim SY, et al. MMPs-specific PEGylated peptide-DOX conjugate micelles that can contain free doxorubicin[J]. Eur J PharmBiopharm, 2007, 67(3): 646-654.
[16] Watson JC, Balster DA, Gebhardt BM, et al. Growing vascular endothelial cells express somatostatin subtype 2 receptors[J]. Br J Cancer, 2001, 85(2): 266-272.
[17] Firer MA, Gellerman G. Targeted drug delivery for cancer therapy: the other side of antibodies[J/OL]. J Hematol Oncol, 2012, 5: 70 [2014-01-13]. http://www.jhoonline.org/content/ pdf/1756-8722-5-70.pdf.
[18] Zhang P, Cheetham AG, Lock LL, et al. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site[J]. Bioconjug Chem, 2013, 24(4): 604-613.
[19] Munyendo LL, Huixia LV, Benza-Ingoula H, et al. Cell penetrating peptides in the delivery of biopharmaceuticals[J/ OL]. Biomolecules, 2012, 2(2): 187-202 [2014-01-13]. http:// www.mdpi.com/2218-273X/2/2/187/pdf.
[20] Sun LC, Coy DH. Somatostatin receptor-targeted anti-cancer therapy[J]. Curr Drug Deliv, 2011, 8(1): 2-10.
[21] Nagy A, Schally AV. Targeting of cytotoxic luteinizing hormone-releasing hormone analogs to breast, ovarian, endometrial, and prostate cancers[J]. Biol Reprod, 2005, 73(5): 851-859.
[22] Keller G, Schally AV, Nagy A, et al. Targeted chemotherapy with cytotoxic bombesin analogue AN-215 can overcome chemoresistance in experimental renal cell carcinomas[J]. Cancer, 2005, 104(10): 2266-2274.
[23] Engel JB, Schally AV, Halmos G, et al. Targeted therapy with a cytotoxic somatostatin analog, AN-238, inhibits growth of human experimental endometrial carcinomas expressing multidrug resistance protein MDR-1[J]. Cancer, 2005, 104(6): 1312-1321.
[24] Szepeshazi K, Schally AV, Block NL, et al. Powerful inhibition of experimental human pancreatic cancers by receptor targeted cytotoxic LH-RH analog AEZS-108[J]. Oncotarget, 2013, 4(5): 751-760.
[25] Jaszberenyi M, Schally AV, Block NL, et al. Inhibition of U-87 MG glioblastoma by AN-152 (AEZS-108), a targeted cytotoxic analog of luteinizing hormone-releasing hormone[J]. Oncotarget, 2013, 4(3): 422-432.
[26] Emons G, Tomov S, Harter P, et al. Phase II study of AEZS-108 (AN-152), a targeted cytotoxic LHRH analog, in patients with LHRH receptor-positive platinum resistant ovarian
cancer[J]. J Clin Oncol, 2010, 28: 5035.
[27] Sun LC, Luo J, Mackey VL, et al. Effects of camptothecin on tumor cell proliferation and angiogenesis when coupled to a bombesin analog used as a targeted delivery vector[J]. Anticancer Drugs, 2007, 18(3): 341-348.
[28] Moody TW, Sun LC, Mantey SA, et al. In vitro and in vivo antitumor effects of cytotoxic camptothecin-bombesin conjugates are mediated by specific interaction with cellular bombesin receptors[J]. J Pharmacol Exp Ther, 2006, 318(3): 1265-1272.
[29] Schally AV, Nagy A. Chemotherapy targeted to cancers through tumoral hormone receptors[J].Trends Endocrinol Metab, 2004, 15(7): 300-310.
[30] Sun LC, Luo J, Mackey LV, et al. A conjugate of camptothecin and a somatostatin analog against prostate cancer cell invasion via a possible signaling pathway involving PI3K/ Akt, alphaVbeta3/alphaVbeta5 and MMP-2/-9[J]. Cancer Lett, 2007, 246(1-2): 157-166.
[31] Sun L, Fuselier JA, Coy DH. Effects of camptothecin conjugated to a somatostatin analog vector on growth of tumor cell lines in culture and related tumors in rodents[J]. Drug Deliv, 2004, 11(4): 231-238.
[32] Kiaris H, Schally AV, Nagy A, et al. A targeted cytotoxic somatostatin (SST) analogue, AN-238, inhibits the growth of H-69 small-cell lung carcinoma (SCLC) and H-157 non-SCLC in nude mice[J]. Eur J Cancer, 2001, 37(5): 620-628.
[33] Engel JB, Schally AV, Halmos G, et al. Targeted cytotoxic bombesin analog AN-215 effectively inhibits experimental human breast cancers with a low induction of multi-drug resistance proteins[J]. Endocr Relat Cancer, 2005, 12(4): 999-1009.
[34] Keller G, Schally AV, Nagy A, et al. Effective therapy of experimental human malignant melanomas with a targeted cytotoxic somatostatin analogue without induction of multi-drug resistance proteins[J]. Int J Oncol, 2006, 28(6): 1507-1513.
[35] Bajo AM, Schally AV, Halmos G, et al. Targeted doxorubicincontaining luteinizing hormone-releasing hormone analogue AN-152 inhibits the growth of doxorubicin-resistant MX-1 human breast cancers[J]. Clin Cancer Res, 2003, 9(10 Pt 1): 3742-3748.
[36] Günthert AR, Gründker C, Bongertz T, et al. Internalization of cytotoxic analog AN-152 of luteinizing hormone-releasing hormone induces apoptosis in human endometrial and ovarian cancer cell lines independent of multidrug resistance-1 (MDR-1) system[J]. Am J Obstet Gynecol, 2004, 191(4): 1164-1172.
[37] Sun LC. The Novel Applications of Anticonvulsant Drug Valproic Acid in Cancer Therapeutics[M]//Boucher A. Valproic Acid: Pharmacology, Mechanisms of Action and Clinical Implications. New York: Nova Science Publishers, Inc., 2012: 1-39.
[38] Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, et al. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors[J]. Cancer Treat Rev, 2008, 34(3): 206-222.
[39] Tsai C, Leslie JS, Franko-Tobin LG, et al. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II[J]. Arch Gynecol Obstet, 2013, 288(2): 393-400.
[40] Franko-Tobin LG, Mackey LV, Huang W, et al. Notch1-mediated tumor suppression in cervical cancer with the involvement of SST signaling and its application in enhanced SSTR-targeted therapeutics[J]. Oncologist, 2012, 17(2): 220-232.
收稿日期:(2014-04-15)
通讯作者:*孙立春, 男,博士,教授,从事多肽药物的研发。E-mail: lsun@tulane.edu
文章编号:1006-1533(2015)01-0069-06
文献标识码:A
中图分类号:R979.1
猜你喜欢
中老年保健(2021年4期)2021-08-22
中国医药导报(2016年29期)2016-12-27
中国实用医药(2016年29期)2016-12-26
上海医药(2016年23期)2016-12-22
哈尔滨医药(2016年3期)2016-12-01
医学信息(2016年29期)2016-11-28
现代检验医学杂志(2016年4期)2016-11-15
癌变·畸变·突变(2016年3期)2016-02-27
西南国防医药(2015年6期)2015-02-28
肿瘤预防与治疗(2015年1期)2015-01-21
