
水及其表面活性剂体系汽—液界面行为的分子动力学模拟
2015-12-31 02:43颜群,王宝和
河南化工 2015年4期
关键词:表面活性剂
水及其表面活性剂体系汽—液界面行为的分子动力学模拟
颜群 , 王宝和
(大连理工大学 化工学院 , 辽宁 大连116024)
摘要:采用分子动力学模拟技术,对水及其表面活性剂体系的汽—液界面行为进行了研究。模拟结果表明,随着温度的升高,纯水体系液相主体密度降低,气—液界面厚度增大,界面张力逐渐减小;水—十二烷基硫酸钠体系与纯水体系相比,汽—液界面厚度明显增大,汽—液界面张力明显减小,其随温度的变化规律和纯水体系一致。
关键词:分子动力学 ; 表面活性剂 ; 汽—液界面 ; 水
中图分类号:TQ423
收稿日期:2015-01-29
作者简介:颜群(1990-),男,在读硕士,从事分子动力学模拟研究,电话:15326179568。
Molecular Dynamics Simulation of Vapor-liquid Interface Behavior for
Water and Surfactant Aqueous Solution Systems
YAN Qun , WANG Baohe
(School of Chemical Engineering, Dalian University of Technology , Dalian116024 , China)
Abstract:The vapor-liquid interface behavior of water and surfactant aqueous solution systems is investigated by molecular dynamics simulation. The simulation results indicate that the vapor-liquid interface thickness of water increases as temperature increases, but the density of liquid bulk and the interface tension show opposite tendency. It is also found that the vapor-liquid interface thickness increases obviously and the interface tension reduces significantly compared to the water system, while its variation with temperature is consistent with water system.
Key words:molecular dynamics ; surfactant ; liquid-vapor interface ; water
0引言
众所周知,表面活性剂具有降低水的表面张力能力,其在气—液界面上的吸附行为是发挥效用的关键。气—液界面热力学行为一直是相变传热传质研究的重点。由于气—液界面厚度非常薄,这就使得其理论分析和实验研究变得十分困难。近些年来,随着计算机技术的迅猛发展,越来越多的学者采用分子动力学(MD)模拟方法,来研究气—液相变界面特性。Kuhn等[1-2]采用分子动力学方法,考查了气—液界面上的脂肪醇聚氧乙烯醚非离子表面活性剂(C12E5)单分子层的结构参数以及分子的动态行为。Wu等[3]采用分子动力学模拟技术,分析了不同种类的胺基Gemini型表面活性剂在正庚烷—水体系的界面张力、密度分布,以及分子的微观结构,其模拟结果与实验吻合良好。苑世领等[4]用分子动力学模拟的方法,研究了阴离子表面活性剂十二烷基硫酸钠(SDS)在汽—液界面上的结构和动力学性质。肖红艳等[5]研究了不同油相和盐度条件下表面活性剂—烷烃—水体系的界面结构,给出了径向分布函数、二面角几率变化等动力学结构信息。本文拟采用分子动力学模拟方法,利用LAMMPS软件模拟水及其表面活性剂体系的气—液界面行为。
1模拟方法
1.1模拟体系
采用直角坐标系,水体系的模拟盒子(初始状态)如图1所示,其大小为Lx×Ly×Lz=12 nm×4 nm×4 nm。液体水分子以面心立方(FCC)晶格方式排列于模拟盒子的中央,汽相分别处于液相的左右两侧,整个模拟体系中有两个气—液界面。

图1 水体系的模拟盒子(初始状态)
采用直角坐标系,水—十二烷基硫酸钠表面活性剂体系的模拟盒子(初始状态)如图2所示,其大小为Lx×Ly×Lz=12 nm×4 nm×4 nm。液体水分子以随机分布的方式位于模拟盒子的中央,两侧各有一相对的表面活性剂单分子层,汽相分别处于液相的左右两侧,整个模拟体系中有两个气—液界面。

图2 水—十二烷基硫酸钠体系的模拟盒子(初始状态)
1.2势能模型
水分子模型很多,如SPC、SPCE、TPI3P和TPI4P等,其结构示意图和模型参数分别见图3和表1[6]。水分子的势能函数如式(1)所示[7-8]。
(1)

图3 不同水分子模型的结构示意图
图3a中为SPC、SPCE和TIP3P模型,b为TIP4P模型(L:负电荷作用点;H:正电荷作用点)

表1 水分子模型参数
表中:q,电量,C;σ,尺度参数,nm;ε,能量参数,J;kB,玻尔兹曼常数,J/K;r,分子间距,nm;θ键角,(°)。
在水—表面活性剂体系的MD模拟中,十二烷基硫酸钠采用全原子模型,力场参数基于AMBER力场,其函数形式如方程(2)所示[9]。
(2)
式中:kr、kθ、Vn分别为键力常数、弯曲力常数、二面角扭曲常数;l0、θ0分别为标准键长和标准键角;n为整数(绕键旋转360°时出现的能量最小值的数目);φ为二面角;rij为原子i和j之间的距离;静电相互作用项中的q表示原子上的电荷数,e。不同原子间的范德华相互作用项中的εij和σij,采用Lorentz-Berthelot混合规则。
1.3模拟细节
水体系模拟在x、y、z方向均采用周期性边界条件,原子间力的截断半径为12 nm,模拟时间步长为1 fs,总模拟时间为0.6 ns,前0.4 ns使得系统达到平衡,后0.2 ns统计计算并输出系统的密度分布、界面张力以及界面厚度。采取正则系综(NVT),并采用Woodcock控温法维持体系温度衡定;依照设定的温度,随机分布分子的初始平动速度;为了保证水分子不偏离盒子中心,每隔1 000步矫正体系的质心,使之在x、y、z方向始终处于盒子的中心处;水—十二烷基硫酸钠体系模拟原子间力的截断半径为10 nm,库伦力的截断半径为12 nm;模拟时间步长为1 fs,总模拟时间为1.4 ns,前1.0 ns使得系统达到平衡,后0.4 ns统计计算并输出数据,其他的模拟设置同水体系一样。本文模拟数据均采用LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)软件计算得到。
2结果与讨论
2.1纯水气—液界面行为的分子动力学模拟
选择SPC、SPCE、TIP3P和TIP4P为水分子模型,分别在300、350、400、450、500和550 K的温度下进行MD模拟,盒内水分子共有1 372个,选择NVT系综,截断半径是12 nm。通过模拟得到纯水体系的密度分布、界面厚度和界面张力。
2.1.1密度分布
不同温度下纯水的初始密度如表2所示。
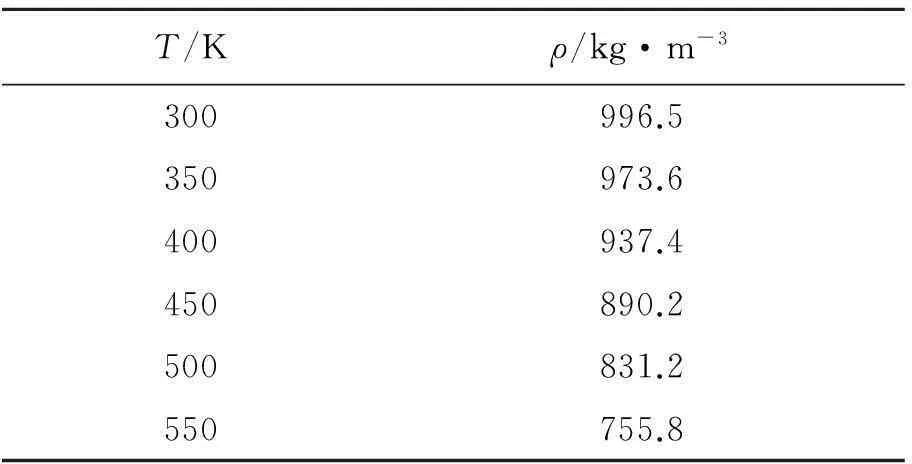
表2 不同温度下纯水的初始密度
模拟分子数N=1 327,四种水分子模型分别在温度T=300、350、400、450、500和550 K时,模拟得到的密度分布如图4所示。从图4可以看出,曲线可以划分为三个部分,分别为汽相主体、液相主体以及气—液界面层。随着温度的增加,液相主体密度逐渐降低,气相主体密度逐渐升高,气—液界面区域逐渐变宽。

(a)SPC (b)SPCE (c)TIP3P (d)TIP4P
将四种水分子模型模拟得到的不同温度下的液相主体密度与实验值比较,如图5所示。由图5可见,四种模型的模拟值和实验值相比都偏低,且温度越高,模拟值与实验值的误差越大;SPCE和TIP4P模型得到的液相密度与实验值的误差较小。
2.1.2界面厚度
根据“10-90”法则进行计算,分别求得SPC、SPCE、TIP3P和TIP4P在300~550 K的界面厚度如图6所示。
从图6中可以看出,随着温度的增加,界面厚度在不断增加,而且温度越高增加幅度越大,其中TIP3P模型的界面厚度增长幅度最大。

图5 不同水分子模型的液相主体密度与实验值比较
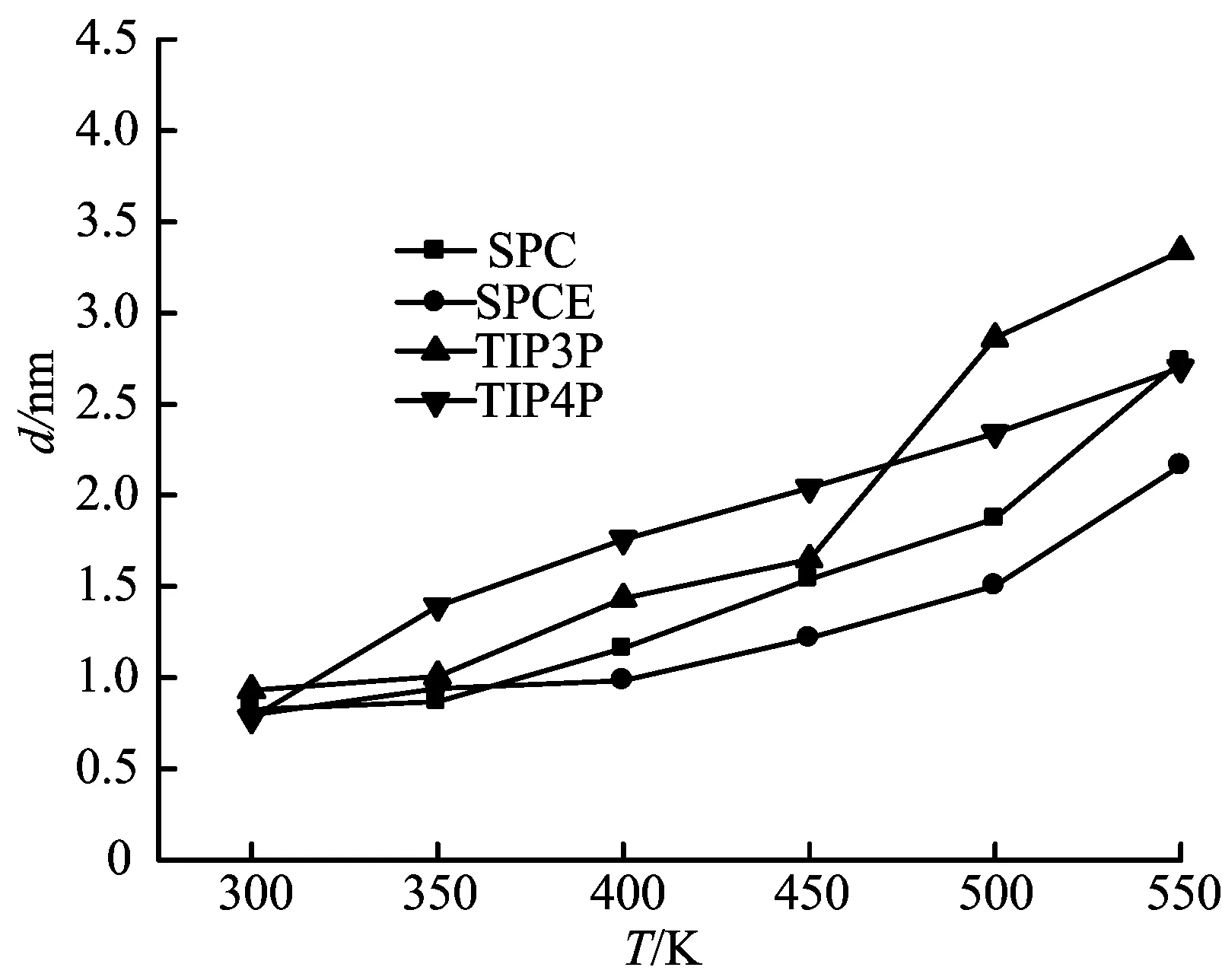
图6 纯水体系的界面厚度
2.1.3界面张力
四种水分子模型分别在温度T=300、350、400、450、500和550 K时,模拟得到界面张力,如图7所示。从图7中可以看出,随着体系温度的升高,界面张力降低,并且模拟值与实验值之间误差逐渐减小。SPCE模型得到的界面张力与实验值的误差较小。
图7 不同水分子模型的界面张力与实验值比较
通过液相主体密度和界面张力的模拟结果可知,SPCE的模拟效果较好,所以在研究水—表面活性剂体系气—液界面行为时,选择SPCE模型。
2.2水—表面活性剂体系气—液界面行为的分子动力学模拟
分别在300、350、400、450、500和550 K温度下进行MD模拟,盒内水分子数为3 000个,两侧的十二烷基硫酸钠数目为10,选择NVT系综,截断半径1 nm,库仑力的截断半径为1.2 nm。模拟得到水—表面活性剂体系的密度分布、界面厚度和界面张力。
2.2.1密度分布
向纯水中加入十二烷基硫酸钠表面活性剂,水分子3 000个,十二烷基硫酸钠20个,模拟得到300 K下水—表面活性剂体系的密度分布,如图8所示。由图8可见,对于只有水的体系来说,其密度变化基本是符合由汽相到液相逐渐增加的趋势,而对于加入的表面活性剂十二烷基硫酸钠的体系来说,其密度的变化情况与只有水的体系有明显的不同。从图8可以看出,在气—液两相的过渡区域,加表面活性剂的体系密度出现明显的增长。

图8 水—表面活性剂体系的密度分布
2.2.2界面厚度
界面厚度取从水相体相密度的90%到表面活性剂体相密度的90%。模拟水分子数N1=3 000,十二烷基硫酸钠数N2=20,在温度T=300、350、400、450、500和550 K时,模拟得到界面厚度,将其与纯水体系的界面厚度对比,如图9所示。从图9可以看出,水—表面活性剂体系的界面厚度随温度的增加而增加,而且和纯水体系的界面厚度对比可知,水—表面活性剂体系的气—液界面厚度明显增大。同时,对纯水体系和水—表面活性剂体系的界面厚度模拟值进行拟合可分别得到式(4)和(5)。
d=-8.620 38+0.050 10T
(4)
d=-8.697 14+0.084 23T
(5)
式中:d为界面厚度,nm;T为温度,K。

图9 水—表面活性剂体系与纯水界面厚度对比
2.2.3界面张力
模拟水分子数N1=3 000,十二烷基硫酸钠数N2=20,温度T=300 K,水—表面活性剂体系的局部界面张力见图10。由图10可知,从汽相主体向液相过渡过程中,界面张力值逐渐增加,在气—液界面区达到峰值;在液相主体又在零值附近波动。

图10 水—表面活性剂体系的局部界面张力
不同温度下的水—表面活性剂体系的界面张力与SPCE模型的界面张力对比如图11所示。
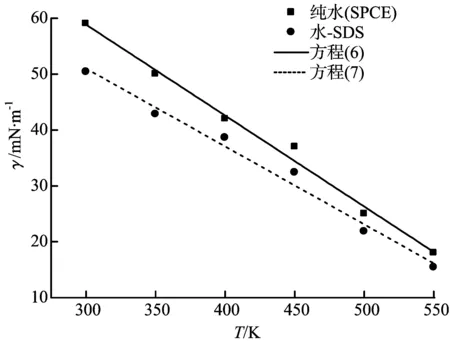
图11 水—表面活性剂体系与纯水的界面张力对比
从图11可以看出,水—表面活性剂体系的界面张力随温度的升高而降低,而且加入十二烷基硫酸钠后水的界面张力明显降低。对纯水体系和水—表面活性剂体系的界面张力模拟值进行拟合可分别得到式(6)和(7)。
γ=107.714 29-0.162 86T
(6)
γ=92.872 380-0.139 54T
(7)
式中:γ为界面张力,mN/m;T为温度,K。
3结论
采用分子动力学模拟技术,对水及其表面活性剂体系的气—液界面行为进行研究。结果表明,随着温度的升高,纯水体系液相主体密度降低,气—液界面厚度增大,张力逐渐减小;SPCE模型与实验值的误差较小;十二烷基硫酸钠—水混合体系与纯水体系相比,气—液界面厚度明显增大,界面张力明显减小,其随温度的变化情况和纯水体系一致。
参考文献:
[1]Kuhn H,Rehage H.Molecular dynamics computer simulations of surfactant monolayers monododecyl pentaethylene glycol at the surface between air and water[J].Journal of Physical Chemistry,B,1999,103(40):8493-8501.
[2]Kuhn H,Rehage H.Molecular orientation of monododecyl pentaethylene glycol (C12E5) surfactants at infinite dilution at the air/water interface.A molecular dynamics computer simulation study[J].Physical Chemistry Chemical Physics,2000,2(5):1023-1028.
[3]Wu R,Deng M,Kong B,et al.Molecular dynamics simulations of ammonium surfactant monolayers at the heptane/water interface[J].Phys Chem B,2009,113: 12680-12686.
[4]苑世领,崔鹏,徐桂英,等.汽液界面上阴离子表面活性剂单层膜的分子动力学模拟[J].化学学报,2006,16(1):1-6.
[5]肖红艳,甄珍,孙焕泉,等.阴离子表面活性剂在水/正烷烃界面的分子动力学模拟[J].物理化学学报,2010,26(02):422-428.
[6]孙杰,何雅玲,李印实,等.水分子模型差异对汽液界面参数分布的影响[J].工程热物理报,2009,30(1):1-4.
[7]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
[8]王蕴博.Mg(OH)2粉体干燥动力学及水分子动力学模拟[D].大连:大连理工大学,2012.
[9]延辉,苑世领. 两亲分子自组装体系及其耐盐机理的理论研究[D].济南:山东大学,2011.
猜你喜欢
现代商贸工业(2017年2期)2017-03-28
江苏农业科学(2016年11期)2017-03-21
科教导刊·电子版(2016年24期)2016-10-29
湖南大学学报·自然科学版(2016年6期)2016-07-14
企业文化·中旬刊(2016年6期)2016-06-16
科技视界(2016年2期)2016-03-30
湖北农业科学(2014年18期)2014-11-20
现代电子技术(2014年8期)2014-09-27
