
HSS和HS反应机理的理论研究
2015-12-29 05:30王智香,曹佳
延安大学学报(自然科学版) 2015年1期
HSS和HS反应机理的理论研究
王智香,曹佳*
(延安大学化学与化工学院,陕西延安716000)
摘要:在B3LYP/6-311+G(3df,2p)水平上对HSS和SH反应过程中所有物种进行了构型优化和频率计算,用高水平G2M(CC5)方法对各个物种做了单点能校正,获得了反应的势能剖面。结果表明,HSS与SH反应体系中主要存在2条反应通道,其中生成P1(H2S+S2)的通道为优势通道,此通道包含2条路径具有相同的决速步,对应表观活化能为8.15 kJ·mol`(-1)。另一条为生成P2(H2+S3)的过程,该反应需克服18.37 kJ·mol`(-1)的表观活化能。利用经典过渡态理论,计算了上述2条通道主路径决速步在200~2000 K温度变化范围内速率常数,此结果可用于后续实验研究。
关键词:HSS;HS;反应机理;速率常数
中图分类号:O641
文献标识码:A
文章编号:1004-602X(2014)04-0021-03
收稿日期:2014-10-19
基金项目:陕西省教育厅专项科研计划项目(2013JK0667);延安大学青年项目资助(YDQ2013-16);延安大学化学与化工学院自然科学专项基金资助(YDHG2014-12)
通讯作者
作者简介:王智香(1983—),女,河北沧州人,延安大学助理实验师。*为
Abstract:The equilibrium geometries and frequencies of all the stationary point for the reaction of HSS with HS are calculated using B3LYP/6-311+G(3df,2p) method.Single point energies of all the species are computed at the G2M(CC5) level,and single potential energy surface is constructed.Three channels (four pathways) are found for the HSS+SH reaction.The result suggested that the formation of P1(H2S+S2) is dominated channel including two pathways,which had the same rate-determining step with the apparent activation energy of 8.15 kJ·mol`(-1).In addition,the product P2(H2+S3) is also confirmed with the apparent activation energy of 18.37 kJ·mol`(-1).The rate constants are evaluated by means of the classical transition state theory over the temperature range of 200~2000 K.The result indicated the rate constant increases with the temperature range from 200 to 2000 K.
煤炭热转化过程中产生的含硫物可腐蚀设备和导致催化剂中毒,硫组分的去除是实现对高硫煤高效利用的关键。通常采用燃前脱硫、燃中固硫和燃后脱硫三个步骤实现去硫[1,2]。由于燃后脱硫步骤硫组分含量相对少,实际操作主要关注前两个脱硫步骤,但是,燃后脱硫步骤未处理掉的含硫物以自由基形式进入大气造成环境污染[3]。研究这些含硫自由基参与的大气反应机理对消除此类含硫污染物具有重要实际意义[4,5]。HSS是煤炭加工过程释放进入大气含量较多的自由基之一。目前已对HSS与H,O,OH和S反应的可能产物和速率常数做了研究。然而,HSS和HS反应也是一个重要过程。Cerru等人发现由HSS和HS反应生成产物为H2S和S2,并未研究其它可能的产物以及反应内在机理[4]。因此,本文从理论上对HSS与SH反应的机理及可能产物和反应速率常数进行了详细研究。
1计算方法
采用B3LYP/6-311+G(3df,2p)方法对所有物种进行构型优化。在相同水平上做了频率分析,用于验证各个物种的本质。根据内禀反应坐标验证过渡态是否连接到正确的反应物和产物。为了获得准确的能量信息,G2M(CC5)方法对各个物种做了高水平单点能校正[6]。上述计算由Gaussian 03程序计算完成[7]。此外,基于传统过渡态理论,应用VKlab程序计算了反应的速率常数[8]。

图1 用B3LYP/6-311+G(3df,2p)方法优化所得的
2结果与讨论
从表1给出的构型参数可以看出,实验与计算的键长和键角值较为接近。例如,H2的计算值为0.0743 nm,文献值为0.0741 nm[9],偏差为0.0002 nm。又如,对于H2S,H2S分子∠H(1)SH(2)理论值为92.4°,S-H键长值为0.1341 nm,而实验∠H(1)SH(2)键角为92.1°,实验S-H键长为0.1336 nm[9],偏差均较小。由此看出,用B3LYP/6-311+G(3df,2p)方法优化的几何构型(如图1)是合适的。

表1 理论值与实验所获键长、键角的比较
2.1 反应机理
图2给出了HSS和HS反应的2条通道(3条路径),分别生成产物H2S+S2和S3+H2。
2.1.1H2S+S2产物
由图2和图3可知,与产物通道P2(S3+H2)相比,由反应物R(HSS+SH)生成到产物P1(H2S+S2)的表观活化能均较小,是主反应通道,该通道包含两条路径Path1和Path2,经过相同的决速控制步。
如图1所示,在Path1中,反应物SH的S原子从侧面进攻SSH的端侧S(2)原子,当二者之间距离为0.2072 nm时,首先形成稳定的中间体IM1。由IM1出发,当SH中的S原子与SSH中的S原子的键长缩短至0.2038 nm,而SSH中的S原子与H原子被缩短至0.1341 nm时,形成过渡态TS1。R(HSS+SH)经过中间体IM1到达过渡态TS1,表观活化能为-148.75 kJ·mol-1。随着反应的不断进行,当S(1)-H(1)键键长为0.1347 nm,两S(2)-S(3)键键长在0.2073 nm时生成中间体IM2,此过程放热30.70 kJ·mol-1。随后IM2中H(1)原子逐渐向H(2)原子方向移动,当H(1)-S(1)变为0.2007 nm,而键角∠H(1)S(1)S(2)为68.6°时反应到达过渡态TS4,TS4对应的表观活化能为8.15 kJ·mol-1,此步为该通道的决速控制步。最终反应经过后中间体IM5生成产物P1(H2S+S2)。
在path2中反应物R(HSS+SH)中的SH中的S(3)原子从中间进攻SSH中的中间S(2)原子,当两原子之间的距离为0.2184 nm时,首先形成稳定的中间体IM3。由IM3出发,发生构型上的转化两H原子逐渐远离,而S原子逐渐靠近H原子,SH中的S原子与SSH中的S原子之间的距离也在逐渐缩短。当两原子距离为0.2070 nm时,形成过渡态TS2,经过中间体IM4和过渡态TS3,到达稳定中间体IM2。TS3形成中间体IM2释放能量186.94 kJ·mol-1。Path1和path2在之后的反应中,同样经过了稳定中间体IM2、过渡态TS4、稳定中间体IM5,最后生成产物P1(S2+H2S)。经过过渡态TS4,表观活化能为8.15 kJ·mol-1,生成的中间体IM5很快转换到产物P1(S2+H2S)。
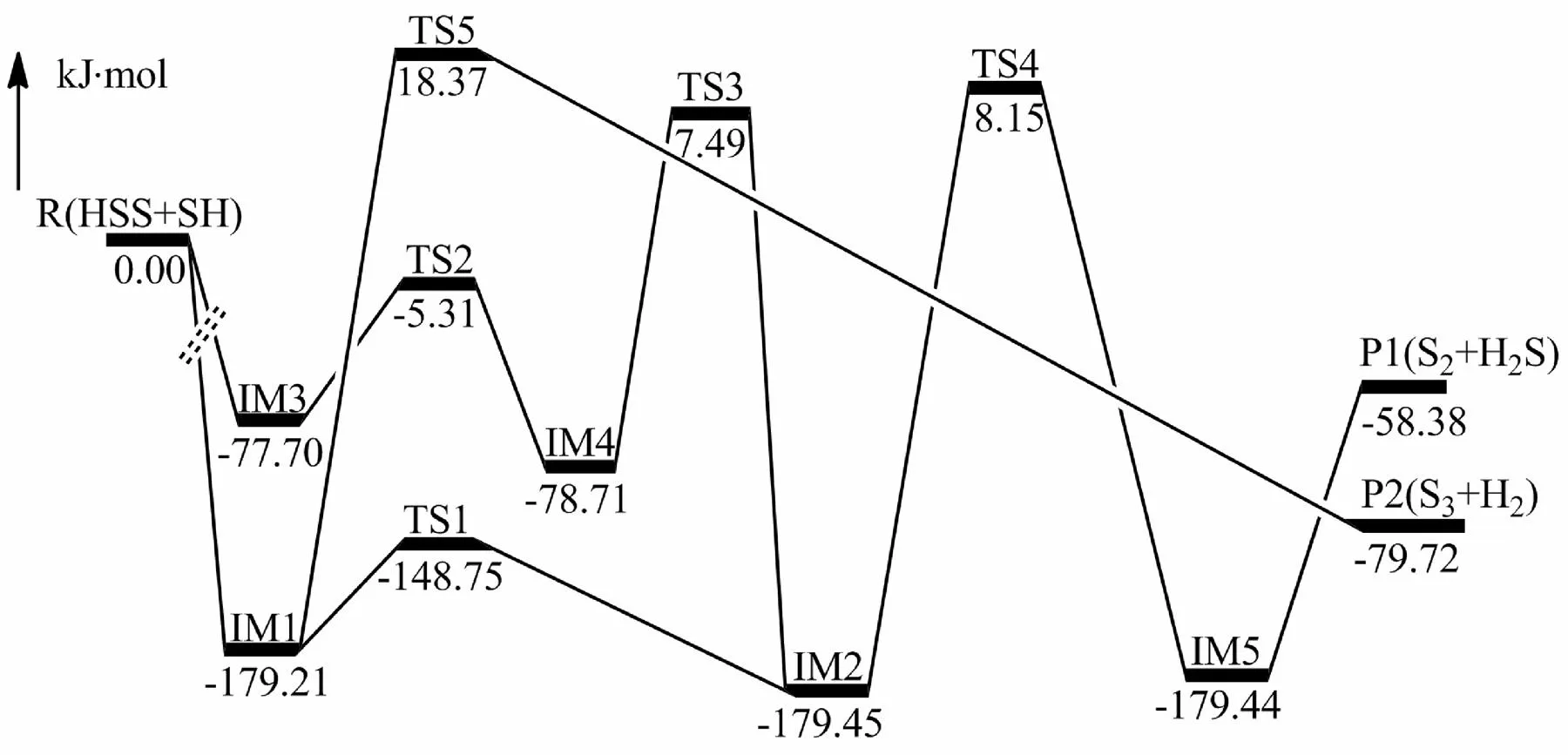
图3 反应HSS和HS在G2M(CC5)//B3LYP/
2.1.2H2+S3产物
R(HSS+SH)→IM1→TS5→P2(S3+H2)
由图2可看出反应物R(HSS+SH)经过path3生成产物P2(S3+H2)。与path1比较,相同的是,SH中的S原子也是进攻SSH中端侧S原子,当两原子之间的距离逐渐缩短,首先形成稳定的中间体IM1。由IM1出发,两端H(1)和H(2)原子逐渐靠近,当∠H(1)S(1)S(2)变为92.0°,而∠H(2)S(3)S(2)变为91.8°,形成过渡态TS5,对应的表观活化能为18.37 kJ·mol-1的表观活化能。随着反应的进行,两H原子结合而远离连接的相应S原子,生成产物P2(S3+H2),放热79.72 kJ·mol-1。相比产物P1(H2S+S2),产物P2(S3+H2)并不是主产物,但是从动力学角度,该通道能垒不高,较易发生,从理论上提供了一种从含硫自由基小分子彼此耦合脱硫且生成H2,具有一定的实际应用价值。

表2 反应决速控制步的速率常数
2.2 决速控制步速率常数
表2列出了分别生成产物P1(H2S+S2)和P2(S3+H2)的决速步速率常数k1和k2。由表2可知,在研究温度范围内,速率常数随着温度的升高而逐渐增大,这也与其具有正的表观活化能相一致,说明高温有利于反应的发生。
3结论
本文采用G2M(CC5)//B3LYP/6-311+G(3df,2p)方法研究了由HSS和HS反应生成产物P1(H2S+S2)和P2(S3+H2)的机理作了研究,P1(H2S+S2)为主产物。利用传统过渡态理论计算了决速步速率常数,发现高温有利于反应发生。
致谢:感谢陕西师范大学理论与计算化学研究室提供计算程序和服务器支持。
参考文献:
[1]沈本贤,程丽华,王海彦,等.石油炼制工艺学[M].北京:中国石化出版社,2009.
[2]陈启文.煤化工工艺[M].北京:化学工业出版社,2009.
[3]刘新宇,张萍,修光利,等.上海市石油化学和化工行业环境污染新问题及监控对策[J].化学世界,2013,9:565-569.
[4]Cerru F G,Kronenburg A,Lindstedt R P.Systematically reduced chemical mechanisms for sulfur oxidation and pyrolysis [J].Combustion and Flame,2006,146: 437-455.
[5]Selim H,Gupta A K,Sassi M.Novel error propagation approach for reducing H2S/O2 reaction mechanism [J].Applied Energy,2012,93: 116-124.
[6]Mebel A M,Morokuma K,Lin M C.Modification of the GAUSSIAN-2 theoretical model: The use of coupled-cluster energies,density-functional geometries,and frequencies [J].J.Chem.Phys,1995,103(17): 7414-7421.
[7]Frisch M J,et al.Gaussian 03,Revision C.02[CP],Gaussian,Inc,Wallingford CT.2004.
[8]Zhang S W,Truong,T.N.VKLab,Version 1.0[CP],University of Utah,Salt Lake City,USA,2001.
[9]Russell D.Johnson Ⅲ.NIST Computational Chemistry Comparison and Benchmark Database[EB/OL].http://cccbdb.nist.gov/,August 2013.
[责任编辑李晓霞]
Theoretical Study on the Reaction Mechanism of HSS and HS
WANG Zhi-xiang,CAO JIA*
(College of Chemistry and Chemical Engineering,Yan'an University,Yan'an 716000,China)
Key words:HSS; SH; reaction mechanism; rate constant
